Article Text

Abstract
Objective Mevalonate kinase (MVK) deficiency is a rare autosomal recessive auto-inflammatory disorder characterised by recurring episodes of fever associated with multiple non-specific inflammatory symptoms and caused by mutations in the MVK gene. The phenotypic spectrum is wide and depends mostly on the nature of the mutations. Hyperimmunoglobulinaemia D and periodic fever syndrome (HIDS) is a relatively mild presentation and predominantly associated with a c.1129G>A (p.V377I) mutation in the MVK gene. We report cases of two sisters homozygous for this mutation but exhibiting distinct (symptomatic vs asymptomatic) phenotypes.
Methods Patient history was obtained; physical and clinical examination and laboratory tests were performed; lipopolysaccharide (LPS) response of peripheral blood mononuclear cells was quantified.
Results Low MVK enzymatic activity is not necessarily associated with inflammatory symptoms. Increased inflammatory cytokine secretion in response to LPS is associated with symptomatic MVK deficiency.
Conclusions Individuals who are homozygous for the common p.V377I mutation in the MVK gene may not display the characteristic inflammatory episodes diagnostic of MKD and thus may be lost for correct and timely diagnosis.
- Disease Activity
- Gene Polymorphism
- Inflammation
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is already known about this subject?
The inflammatory phenotypic spectrum of patients carrying mutations in the Mevalonate kinase (MVK) gene is wide and depends mostly on the nature of the mutations. HyperImmunoglobulinemia D and periodic fever Syndrome (HIDS) is a relative mild presentation and predominantly associated with a c.1129G>A (p.V377I) mutation in the MVK gene.
What does this study add?
This study describes two sisters carrying the same homozygous c.1129G>A mutation but exhibiting distinct (symptomatic versus asymptomatic) phenotypes. This suggests the existence of modifiers loci involved in the penetrance of HIDS.
How might this impact on clinical practice?
Individuals homozygous for the common p.V377I form of the MVK protein may not display the characteristic inflammatory episodes diagnostic for Mevalonate kinase deficiency (MKD) and thus may be lost for correct and timely diagnosis.
Introduction
Mevalonate kinase deficiency (MKD) is a rare autosomal recessive autoinflammatory disorder characterised by recurring episodes of high fever associated with multiple non-specific inflammatory symptoms including headache, arthritis, abdominal pain, diarrhoea and skin rash.1 The disorder is caused by biallelic mutations in the MVK gene and can present with variable clinical phenotypes.2 ,3
The severe presentation is also known as mevalonic aciduria (MA), and includes, in addition to inflammatory episodes, developmental delay, dysmorphic features, ataxia, cerebellar atrophy, psychomotor retardation and possibly premature death. The mild presentation is known as hyperimmunoglobulinaemia D and periodic fever syndrome (HIDS) and characterised predominantly by recurrent fever with inflammatory symptoms.4 Phenotypic overlap between HIDS and MA provides evidence of a phenotypic continuum between both diseases,5 which therefore currently are considered the mild and severe end of the MKD spectrum.
During the inflammatory crises, high serum levels of pro-inflammatory cytokines such as interleukin (IL)-1β, tumour necrosis factor (TNF-α) and IL-6 are observed in patients.6 Their role in the occurrence of fever is suggested by the favourable therapeutic effect of the TNF-α inhibitor etanercept.7 IL-1β is the prototypical proinflammatory cytokine and plays an important role in the pathogenesis of several autoinflammatory diseases, including tumour necrosis factor-1 receptor-associated periodic syndrome and cryopyrin-associated periodic syndromes. IL-1β is produced on pro-IL-1β cleavage by caspase-1, a process that requires activation of inflammasomes, which are responsible for the activation of caspase-1.8 It has been established that the MKD-dependent inflammation results from a shortage of the isoprenoid geranylgeranyl-PP.9 Furthermore, it has also been shown that unprenylated RhoA is involved in IL-1β hypersecretion in the MKD deficiency model through stimulation of Rac1 activity. A pivotal role for RhoA in MKD-associated autoinflammation through Rac1/PKB signalling toward interleukin 1β production has been identified, which partly clarifies the link between MVK deficiency and increased IL-1β secretion.10
We report the cases of two sisters, both homozygous for the most common V377I mutation causing HIDS, but of whom only one exhibited inflammatory symptoms.
Case report
Clinical description of the patient
The proposita (patient 1), a 43-year-old woman with a history of depression and ophthalmic abnormalities (optic neuritis) was examined in the rheumatology clinic because of polyarthralgias involving the fingers, wrists and ankles, and disturbed night sleep. She was married and had two children who had not presented any medical problems up to the time of her examination. The patient reported of diffuse pain concerning joints and muscles but clinical examination revealed absence of synovitis and neurological examination was normal. She presented central allodynia and hyperalgaesia. She also reported of visual field alteration. Other symptoms included frequent right upper abdominal pains, nausea, vomiting and fatigue. She presented no sign of autoimmune diseases: no Raynaud syndrome, no dry syndrome, no photosensitivity, no thromboembolic episode, no miscarriage, no nettle rash, no other inflammatory cutaneous lesions, no fever or other systemic manifestation, no headache and no chest pain. Yet, small cervical infracentimetric painless lymphadenopathies were present.
On the first evaluation, blood cell count was normal, no inflammatory syndrome was found, C reactive protein level was below 4 mg/L, antinuclear antibodies titre was 1/320 and rheumatoid factors were not detected. HIV, viral hepatitis (HBV, HCV) and Lyme disease serologies were negative. In contrast, serum protein immunofixation electrophoresis revealed abnormal levels of polyclonal IgD, which was quantitatively confirmed by nephelometry (690 UI/mL, table 1). IgA levels were quantified at 5.82 U/mL. CT of the chest, abdomen and pelvis was normal, in particular, neither splenomegaly nor adenopathy was observed. Ophthalmological examination and visual evoked potential recording revealed conduction troubles of the right eye optic nerve. At this stage, prescription of low-dose corticoids (20 mg/day) later supplemented with methotrexate (10 mg/week) did not show any efficacy. Despite the absence of (periodic) fever, mutations in the MVK gene encoding mevalonate kinase were searched. To this end, exons and intron/exon junctions of the MVK gene were amplified and sequenced. This revealed the homozygous presence of the c.1129G>A (p.V377I) mutation, which is found in >90% of patients with the HIDS phenotype. The patient was then placed under anti IL-1 therapy (kineret 100 mg/day), which provided a subjective improvement (reduction of the joint pain), with amelioration of neither the ophthalmic manifestation nor the adenopathies.
Clinical and biological features of the proposita and family members
Biochemical studies
For these studies, blood samples and skin biopsies were obtained from patients and controls attending the Department of Rheumatology, Hôpitaux Civils de Colmar (France), and were collected during routine clinical procedures. All participants gave their informed consent and the study was approved by the Ethics Committee of Strasbourg University Hospital.
Because the patient did not exhibit recurrent symptoms and showed neither fever nor biological inflammatory signs, testing for MVK activity in circulating lymphocytes was performed using a radiometric assay based on the quantification of the phosphorylated products of 14C-mevalonic acid, as previously described.11 In addition, urinary mevalonic acid extraction was performed by gas phase chromatography coupled with mass spectrometry. MVK enzymatic activity in lymphocytes was drastically reduced to 0.3 µkat/kg prot (2.5< normal range <7.8 μkat/kg) and urinary mevalonic acid concentration was found twice above normal values (3.1 and 4.1 mmol/mol creatinine). Based on the clinical presentation and combined laboratory results, HIDS was diagnosed.
The atypical clinical presentation of HIDS without fever and other recurrent inflammatory symptoms prompted us to investigate the whole family. The parents were not consanguineous and their ethnic background was Caucasian. The patient had one sister and one brother (figure 1). Table 1 summarises the biological parameters of four family members; the brother was not available for follow-up. Interestingly, the sister, who was 40 years old, had no history of disease and no inflammatory signs, but carried the same homozygous p.V377I mutation, had a similar decrease in MVK enzyme activity in lymphocytes (0.3 µkat/kg prot), and elevated serum levels of IgD (140 U/mL) and IgA (2.5 U/mL). In light of these results, thorough clinical examination was performed, but no symptoms compatible with HIDS were found (no abdominal pain, no vomiting, no aphthous stomatitis, no maculopapular rash, no recurrent headache, no myalgia and no lymph node enlargement).
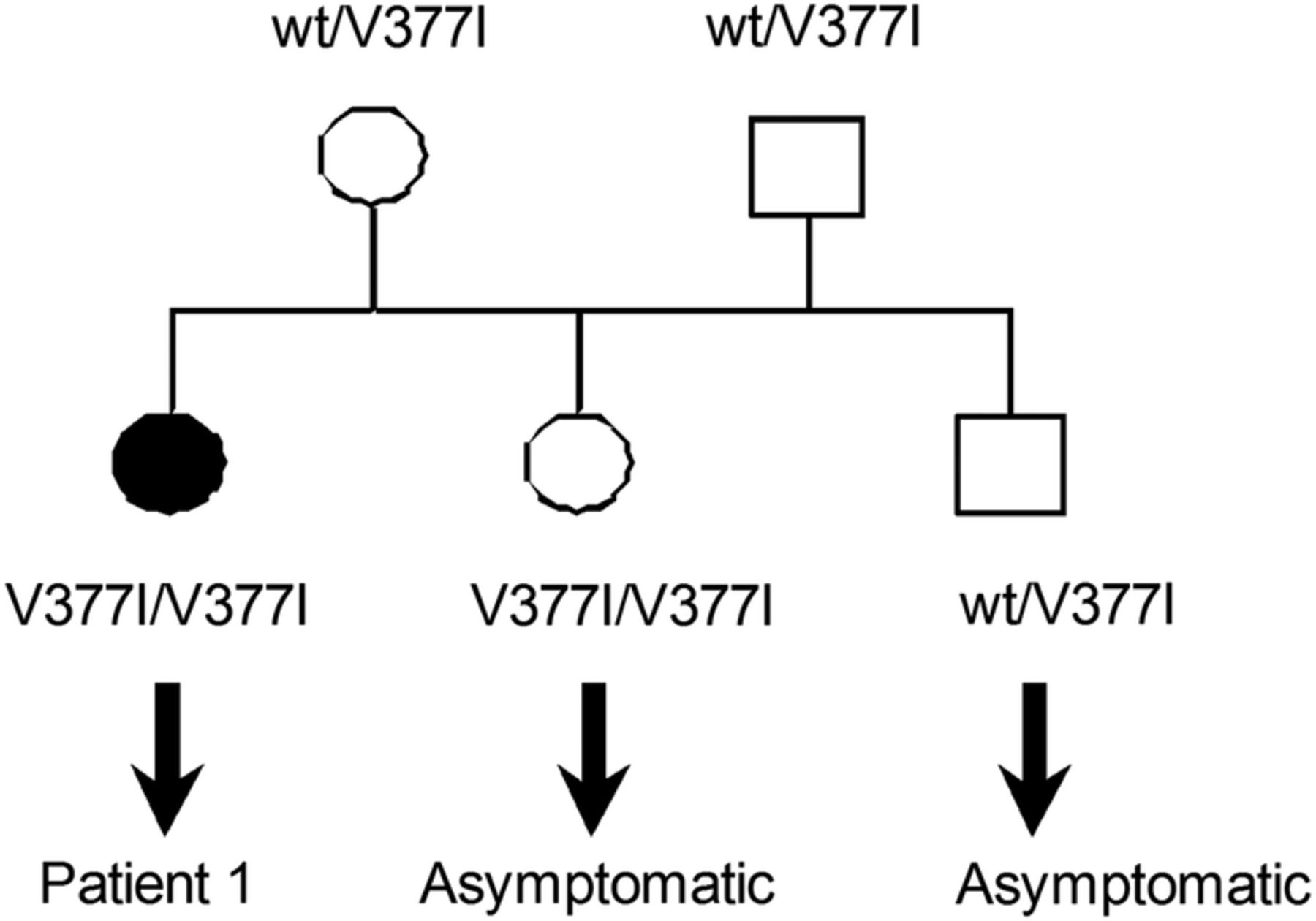
Pedigree of the HIDS family. The black circle corresponds to the symptomatic patient carrying the homozygous mutation. Squares correspond to males. wt, wild-type allele. HIDS, hyperimmunoglobulinaemia D and periodic fever syndrome.
The father (67 years old) had a history of ischaemic cardiopathy and chronic bronchopulmonary obstructive disease, but never experienced recurrent fever, arthritis or other systemic symptoms. Genetic analysis confirmed carriership for the V377I mutation. In accordance with this, the MVK activity in lymphocytes and the urinary mevalonic acid concentration were normal, and serum IgD and IgA levels were low. The mother (69 years old and asymptomatic) was also a carrier for the V377I mutation and showed normal urinary mevalonic acid concentrations, and low serum IgD and IgA levels. No inflammatory signs were detected.
To further investigate the effect of the mutation on MVK enzyme activity, we analysed fibroblasts obtained from skin biopsies of the different family members and controls, and cultured in Nutrient Mixture Ham’ s F-10 with L-glutamine and 25 mM HEPES supplemented with 10% fetal calf serum at room temperature. MVK activity was measured in fibroblast cell lysates using C-labelled mevalonate (NEN, Perkin Elmer) as previously described.12
In accordance with their carrier status and as previously documented (10), the heterozygous parents showed MVK activities that comprise ∼30% of the activities in control fibroblasts. The MVK activities measured in the cells of the patient and her asymptomatic sister comprised 3% and 6.5%, of the activities in control fibroblasts, which are both within the range normally observed for patients with HIDS (10).
Pro-inflammatory cytokine response of lipopolysaccharide (LPS)-stimulated peripheral blood mononuclear cells
We investigated the cytokine response of peripheral blood mononuclear cells (PBMCs) isolated from different members of this family to evaluate if these were different. PBMCs were stimulated with LPS (1 μg/mL, Salmonella abortus equi LPS from Sigma) for 6 h and cytokine (IL-6, IL-1β, TNF-α) release was measured by ELISA (R&D Systems) in culture supernatants. LPS-stimulated PBMCs from the symptomatic patient 1 released large amounts of IL-6 (27 000 pg/mL) while those of her asymptomatic sister (subject 1) released lower quantities of this cytokine (18 000 pg/mL, figure 2A). Similar observations were made for TNF-α (figure 2B) and IL-1β (figure 2C). Altogether, these results indicate differential inflammatory responses in cells isolated from the HIDS symptomatic subject compared to her healthy sister. In this assay, values represent the mean±SD. Two-tailed Mann-Whitney test was used to compare two independent groups using GraphPad software. A probability (p) value <0.05 was considered significant.

{kind=link}
{kind=link}
Proinflammatory cytokine production by LPS-activated PBMC from patient 1 (symptomatic), patient 2 (asymptomatic), patient 3 (asymptomatic) and two unrelated controls. Cells were stimulated with LPS (1 μg/mL) for 6 hours. (A-C) Cytokine levels in the cell culture supernatant were quantified by ELISA. Values are the mean of 3 experiments±SD. *p<0.05; **p<0.01 (Mann-Whitney U test). Statistical analysis comparing P1 and other participants or controls gave similar results. PBMC, peripheral blood mononuclear cells.
Discussion
In this report, we describe two sisters, aged 43 and 40 years, who were both homozygous for the p.V377I mutation in the MVK gene, which is found in 90% of HIDS patients. The vast majority of these are compound heterozygous for this mutation and a second severe mutation, however, and homozygotes are rarely found, based on which it had been hypothesised previously that a majority of homozygotes may be asymptomatic or present with a less severe inflammatory phenotype.13 Our findings now confirm this hypothesis. Indeed, although biochemical studies in cells from both sisters revealed markedly decreased MVK activities that were within the range typically observed in patients with HIDS, only one of the sisters showed clinical symptoms, including arthralgia, optical neuritis and lymphadenopathies, while the other sister was asymptomatic. Moreover, the symptomatic sister never presented with recurrent episodes of fever, which is considered a diagnostic hallmark of MKD, including in HIDS.14 In both sisters, increased levels of IgD were observed, which is compatible with but not diagnostic, per se, for HIDS. This observation is in line with another recent report describing absence of symptoms in participants carrying a homozygous p.V377I mutation.15 However, in the latter case, asymptomatic children were also carriers for MEFV mutations p.E148Q, p.P369S and p.R408Q. In addition, our work strengthens recent observations indicating that MKD, while very unlikely in patients with a normal mevalonic acid excretion, cannot be excluded completely.16
Homozygosity for the pV377I mutation does result in an augmented pro-inflammatory response of LPS-stimulated PBMCs when compared to control PBMCs. Interestingly, we noted that the TNF-α, IL-6 and IL1-β secretion by cells isolated from patient 1 was higher than observed for her asymptomatic sister (patient 2), her mother (patient 3) or 2 genetically unrelated controls, which may reflect a higher risk for developing inflammation or a pre-primed immunological status of the patient. The father (patient 4) was not tested in this assay because of its elevated erythrocyte sedimentation rate (ESR, table 1) at the time of blood sampling.
To conclude, our report indicates that individuals who are homozygous for the common p.V377I mutation may not display the characteristic inflammatory episodes diagnostic for MKD. Such patients who do not follow the gold standard with respect to clinical presentation14 could thus be lost for correct and timely diagnosis.
To gain further insights into the molecular basis underlying the phenotypic discrepancies between patients 1 and 2 and possibly identify genetic modifiers, further studies involving multi-OMICS analyses—including whole exome sequencing, transcriptomic and proteomic analysis—are ongoing.
Acknowledgments
The authors thank the patients and their family members who participated in this study.
References
Footnotes
Contributors LM, GA, HRW and ND-Y performed experiments and analysed data. LM, GA, PG, RC, HRW, ND-Y, SB and JS analysed data and results. LM, PG, HRW, SB and JS wrote the paper.
Funding This work was funded by the Laboratoire d'Excellence (LABEX) TRANSPLANEX [ANR-11-LABX-0070_TRANSPLANTEX]. Additional support was received from the Institut national de la santé et de la recherche médicale (INSERM), the University of Strasbourg (UNISTRA) and the Institut Universitaire de France (IUF).
Competing interests None declared.
Patient consent Obtained.
Ethics approval Human cells were obtained after informed consent was obtained from donors. Ethical approval was granted by the institutional ethics committee, Hopitaux Civils de Colmar, France.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Data reported in this manuscript will be shared if needed.