Article Text

Abstract
Objectives To evaluate the effect of baricitinib on patient-reported outcomes (PROs) in patients with active rheumatoid arthritis (RA) and an inadequate response or intolerance to conventional synthetic disease-modifying antirheumatic drugs.
Methods In this phase III study, patients were randomised 1:1:1 to placebo (N=228), baricitinib 2 mg once daily (QD, N=229) or baricitinib 4 mg QD (N=227). PROs included the Health Assessment Questionnaire-Disability Index (HAQ-DI), Patient's Global Assessment of Disease Activity (PtGA), patient's assessment of pain, measures from patient electronic daily diaries (duration and severity of morning joint stiffness (MJS), Worst Tiredness, Worst Joint Pain), Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-F), SF-36, EuroQol 5-D index scores and visual analogue scales (VAS) and the Work Productivity and Activity Impairment Questionnaire-RA. The primary time point for the study was week 12. Treatment comparisons were assessed with logistic regression for categorical measures and analysis of covariance for continuous variables.
Results Statistically significant improvements were observed for both baricitinib groups versus placebo in HAQ-DI, PtGA, pain, daily diary measures, EuroQoL index scores and SF-36 physical component score at week 12 and for those measures when assessed at week 24. Baricitinib 2 mg and baricitinib 4 mg were statistically significantly improved versus placebo for the EuroQoL VAS and FACIT-F, respectively, at week 24.
Conclusions Baricitinib 2 or 4 mg provided significant improvement versus placebo in PROs across different domains of RA, including physical function, MJS, fatigue, pain and quality of life.
Trial registration number NCT01721057; Results.
- DMARDs (synthetic)
- Patient perspective
- Rheumatoid Arthritis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Patient-reported outcomes are obtained directly from patients, reflecting how they feel and function in relation to rheumatoid arthritis (RA) and to RA therapy; this information may improve the quality of patient care and health-related quality of life, which is an important treatment goal.
In the RA-BUILD study, baricitinib (2 or 4 mg once daily) was associated with clinical improvement and inhibition of progressive radiographic joint damage in patients with RA and an inadequate response or intolerance to conventional synthetic disease-modifying antirheumatic drugs.
What does this study add?
This study describes the patient-reported outcome (PRO) data collected in RA-BUILD and assesses whether the overall efficacy of baricitinib is reflected in clinically meaningful changes in PROs.
Once-daily baricitinib 2 or 4 mg produced significant improvements compared with placebo in most of the prespecified PROs, including physical function, fatigue, duration and severity of morning joint stiffness, pain and health-related quality of life; for some PROs, including selected daily diary measures, baricitinib treatment with the 4 mg dose resulted in more rapid improvement compared with the 2 mg dose.
How might this impact on clinical practice?
The efficacy of baricitinib is not limited to physician-based or laboratory-based assessments, but translates to a positive benefit with respect to patients' health-related quality of life and overall function; the study results suggest that baricitinib is a potentially valuable addition to the rheumatoid arthritis treatment arsenal for patients struggling with this common and disabling condition.
Introduction
Rheumatoid arthritis (RA) is a chronic, systemic disease associated with inflammatory activity and joint damage that result in disability, pain and other impairments.1–4 Subsequently, RA adversely affects patients' daily activities and health-related quality of life (HRQOL),5–8 and the patient-reported burden of RA is considered an important factor in the management of RA.9 ,10
Selected patient-reported outcomes (PROs) are incorporated in the American College of Rheumatology (ACR) improvement criteria and composite measures of disease activity, such as the Disease Activity Score (DAS), Clinical Disease Activity Index (CDAI) and Simplified Disease Activity Index (SDAI).11 The information obtained with these measures is useful to assess efficacy, yet is limited in assessing the range of health domains and treatment effectiveness important to patients with RA, such as the HRQOL, fatigue, and well-being in daily life and in work. A number of PRO instruments have therefore been developed to measure the physical, emotional and social burden of RA. Since these PRO measures are obtained directly from the patients and are not influenced by other individuals, they may more accurately reflect how the patient feels and functions in relation to RA and to therapy.12 ,13 The PRO measures also may facilitate doctor–patient communication and shared decision-making to improve the quality of patient care,14 ,15 which is an overarching principle recommended by EULAR.16
Baricitinib is a selective inhibitor of Janus kinase (JAK)1/JAK2 that interrupts signalling in pathways for several cytokines considered important in RA pathogenesis.17 In the Phase III RA-BUILD clinical trial, baricitinib (2 or 4 mg once daily (QD)) was associated with clinical improvement and inhibition of progression of radiographic joint damage in patients with RA and an inadequate response (IR) or intolerance to conventional synthetic disease-modifying antirheumatic drugs (csDMARDs).18 The current paper describes the PRO data collected in RA-BUILD and assesses whether the overall efficacy of baricitinib is reflected in clinically meaningful changes in PROs.
Patients and methods
Patients
RA-BUILD (NCT01721057) was a randomised, 24-week, double-blind, placebo-controlled, parallel-group, international phase III study. The primary end point of the study was the proportion of patients achieving an ACR 20% response at week 12 (baricitinib 4 mg vs placebo). Full details of the study have been reported previously.18 Briefly, patients were randomly assigned (1:1:1) to placebo, baricitinib 2 mg or baricitinib 4 mg QD in addition to any stable background therapies. Patients were ≥18 years with active RA (≥6/68 tender and ≥6/66 swollen joints; serum high-sensitivity C-reactive protein ≥3.6 mg/L (upper limit of normal 3.0 mg/L)) and an IR (despite prior therapy) or intolerance to ≥1 csDMARDs. The use of up to two concomitant csDMARDs was permitted, but not required, at study entry; these must have been used for at least the preceding 12 weeks with stable doses for at least the preceding 8 weeks. The study was conducted in accordance with the ethical principles of the Declaration of Helsinki and Good Clinical Practice guidelines and was approved by the institutional review board or ethics committee for each centre involved. All patients provided written informed consent.
Methods
The PROs were prespecified secondary outcomes of the study. Physical function was measured using the Health Assessment Questionnaire-Disability Index (HAQ-DI).19 ,20 Scores range from 0 to 3, with lower scores reflecting better physical function and, thus, less disability. Changes in the HAQ-DI score were assessed in the context of a minimum clinically important difference (MCID) of 0.22.21 Disease activity and arthritis pain were measured using the Patient's Global Assessment of Disease Activity (PtGA) and patient's assessment of pain visual analogue scales (VAS, 0–100 mm); higher scores indicate more disease activity or pain.
Duration of morning joint stiffness (MJS), MJS severity, Worst Tiredness and Worst Joint Pain (referred to as diary PROs) were collected using a daily electronic diary from day 1 through week 12 and were key secondary endpoints. The latter three measures were assessed with numeric rating scales, ranging from 0 to 10, with 10 being the worst level.
Fatigue was assessed by the Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-F) scale (range 0 to 52), with higher scores representing less fatigue.22 For the FACIT-F, a 3–4-point change has been considered an MCID,22–24 and in this study, a value of 3.5623 was used to assess the clinical relevance of changes in FACIT-F scores.
Health-related quality of life (HRQOL) was evaluated using the Medical Outcomes Study (MOS) Short-Form-36 (SF-36; V.2, Acute),25 ,26 in which eight domains are normalised (scored from 0 to 100) and are z-transformed to calculate the physical (PCS) and mental (MCS) component scores. An MCID of 5 was used to assess the clinical relevance of changes in SF-36 component scores.27 ,28
The EuroQol 5-Dimensions (EQ-5D) Health State Profile was also used to assess HRQOL. The EQ-5D consisted of two components: a descriptive system of the respondent's health and a rating of his/her current health state (0–100 mm VAS), in which the end points are labelled ‘best imaginable health state (100)’ and ‘worst imaginable health state (0)’.29 The UK and US scoring algorithms provide an index score using the UK or US population weighting to normalise it to that population;30–32 index score ranged from 0 (death) to 1 (perfect health).
The Work Productivity and Activity Impairment Questionnaire-Rheumatoid Arthritis (WPAI-RA) scale was used to measure the health and symptoms of overall work productivity and impairment of regular activities during the past 7 days. Scores are calculated as impairment percentages with higher percentages indicating greater impairment and less productivity.33
Non-diary PROs were assessed at baseline and at weeks 1, 2, 4 and every 4 weeks thereafter to week 24, with the exceptions of the WPAI-RA (not collected at week 1) and SF-36, EQ-5D and FACIT-F data, which were collected at baseline, week 4 and then every 4 weeks until week 24.
Statistical analyses
Randomised patients treated with ≥1 dose of the study drug were included in the efficacy analyses under a modified intention-to-treat principle (mITT analysis set).
Treatment comparisons for categorical and continuous efficacy measures were performed using logistic regression and analysis of covariance (ANCOVA), respectively, with baseline value (for continuous measures), treatment, geographical region and centrally-confirmed presence of baseline joint erosions in the model. For diary PRO data, the ANCOVA models were based on the average of scores collected in the 7 days prior to the week 12 visit date without baseline adjustment for the duration of MJS and were analysed by the non-parametric method, Wilcoxon rank-sum test. For the ANCOVA model, the least squares means (also considered as estimated marginal means) in each treatment group were derived to estimate the treatment difference. For the non-parametric method, the medians in each treatment group were described. In post hoc analyses evaluating the kinetics of initial effect, daily scores from the day of randomisation (day 1) up to day 28 were also assessed, without weekly averaging. For the daily score analysis, mixed models for repeated measures with treatment, geographical region, centrally confirmed presence of baseline joint erosions (yes/no), study day and the interaction of treatment-by-day as covariates were applied, with duration of MJS, analysed by non-parametric methods using the Wilcoxon rank-sum test. All statistical analyses were conducted using SAS V.9.2 (SAS Institute, Cary, North Carolina, USA).
Patients who were rescued or discontinued were defined thereafter as non-responders (non-responder imputation) for all categorical efficacy measures. For continuous measures, the last observations before rescue or discontinuation were carried forward (modified last observation carried forward method). The WPAI-RA measures were censored after rescue or discontinuation without imputation applied.
For the ANCOVA analysis for continuous variables, t-tests were performed to test the treatment differences. For the logistic regression analyses for categorical variables, Wald tests were performed to assess treatment comparisons. All analyses were based on a significance level of 0.05 (two-sided). p-Values were not adjusted for multiple comparisons.
Results
Patients
A total of 684 patients were randomised: 228 received placebo, 229 received baricitinib 2 mg and 227 received baricitinib 4 mg. Patient disposition has been described in Dougados et al.18 In summary, baseline patient characteristics and disease activity were similar among the groups (table 1). Overall, 298 (44%), 210 (31%) and 171 (25%) patients had previously received 1, 2 or ≥3 csDMARDs, respectively. Baseline PROs indicated a significant disease burden, which was consistent with the baseline clinical disease activity (table 1).
Patient characteristics, disease activity and patient-reported outcomes at baseline
Patient-reported outcomes
HAQ-DI, PtGA and pain
As reported in Dougados et al,18 statistically significant improvements compared with placebo were observed at week 12 for HAQ-DI, PtGA and pain. Furthermore, for baricitinib 4 mg versus placebo, statistically significant improvements were evident as early as week 1 for HAQ-DI (p≤0.05 for all time points), PtGA (p≤0.01 for all time points) and patient's assessment of pain (p≤0.05 for all time points). For baricitinib 2 mg, statistically significant improvements versus placebo were observed as early as week 8 for HAQ-DI (p=0.001 for time points after week 8), week 2 for PtGA (p≤0.05 for time points after week 2) and week 4 for patient's assessment of pain (p≤0.01 for time points after week 4). Significant improvements in physical function and reductions in PtGA and pain were maintained at weeks 12 and 24 (table 3).
The proportion of patients who met or exceeded the MCID for HAQ-DI was 54%, 69% and 64% for placebo, baricitinib 2 mg and baricitinib 4 mg, respectively (p=0.001 for baricitinib 2 mg vs placebo; p=0.027 for 4 mg vs placebo) at week 12 and was 42%, 64% and 60% at week 24 (p=0.001 for both baricitinib groups vs placebo).
Dougados et al18 also assessed treatment effect based on background csDMARD use. A subgroup analysis with the outcome measure, change from baseline in HAQ-DI, suggested no heterogeneity of treatment effect based on background csDMARD therapy (see online supplementary table S1).
supplementary data
Diary PROs: duration of MJS, MJS severity, Worst Tiredness and Worst Joint Pain
At week 12, reductions were observed in duration and severity of MJS (table 2; for duration and severity: p=0.002 for baricitinib 2 mg vs placebo, p=0.001 for baricitinib 4 mg vs placebo), Worst Tiredness (p=0.049 for baricitinib 2 mg vs placebo, p=0.027 for baricitinib 4 mg vs placebo) and Worst Joint Pain (p=0.001 for both baricitinib groups vs placebo).
Day 1, week 1 and week 12 data from patient daily diaries
With baricitinib 4 mg versus placebo, a statistically significant reduction in the severity of MJS (p≤0.05) and in Worst Joint Pain (p≤0.01) was first seen at week 1, in Worst Tiredness at week 2 (p≤0.05) and in duration of MJS at week 4 (p≤0.05).18 With baricitinib 2 mg, the statistically significant (p≤0.05) reductions were first observed at week 8 for the duration and severity of MJS, at week 12 for Worst Tiredness and at week 2 for Worst Joint Pain.18
Consistent with the weekly averaged data, the daily diary scores showed significant improvement in patients receiving baricitinib 2 or 4 mg compared with placebo. The greatest rapidity and magnitude of benefit was observed with baricitinib 4 mg, with improvements observed as early as day 3 for Worst Tiredness, day 4 for the severity of MJS and Worst Joint Pain and day 10 for the duration of MJS (see online supplementary figure S1).
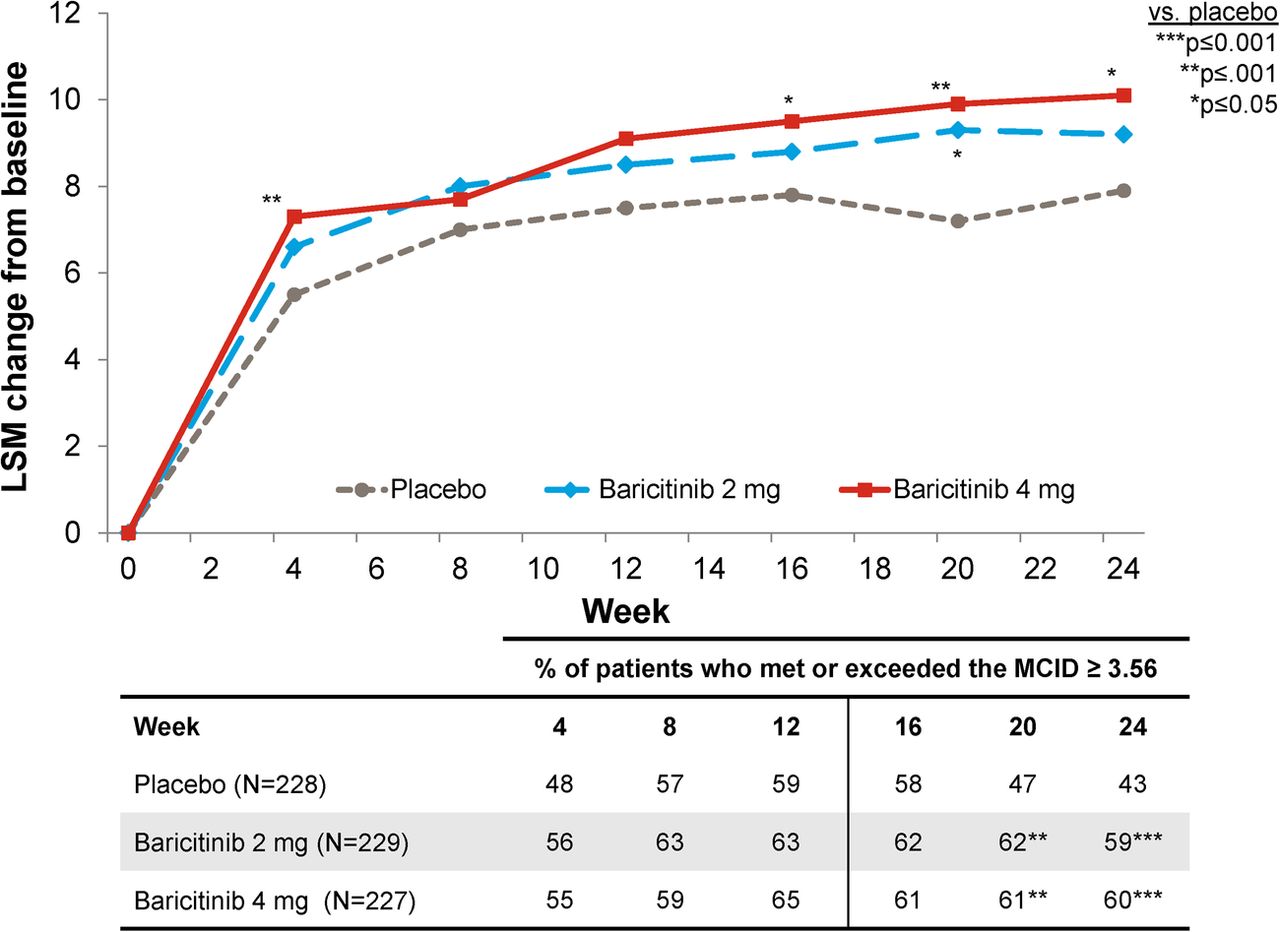
Functional Assessment of Chronic Illness Therapy-Fatigue
Treatment with baricitinib 4 mg was associated with significant improvement in FACIT-F at week 24 (figure 1). The improvements in the FACIT-F score were observed at week 4, the first assessment of the measure (figure 1). Numeric improvements in FACIT-F were observed for baricitinib 2 mg versus placebo, but a statistically significant improvement was only observed at week 20.

Change from baseline for Functional Assessment of Chronic Illness Therapy-Fatigue.
For FACIT-F, the proportion of patients who met or exceeded the MCID was 59%, 63% and 65% for placebo, baricitinib 2 mg and baricitinib 4 mg, respectively, at week 12 (p=0.323 for baricitinib 2 mg vs placebo and p=0.209 for baricitinib 4 mg vs placebo), and was 43%, 59% and 60% at week 24 (p=0.001 for both baricitinib groups vs placebo).
Health-related quality of life
Short Form 36
Patients treated with baricitinib 2 or 4 mg reported statistically significant improvements compared with placebo in most of the 8 SF-36 domains at weeks 12 and 24. The values for the social functioning, role emotional and the mental health domains improved for the baricitinib treatment groups, but differences from placebo were not statistically significant (see online supplementary table S2).
Compared with placebo-treated patients, patients in both baricitinib treatment groups reported statistically significantly improved values for the SF-36 PCS (figure 2A), from the first postbaseline assessment at week 4 and maintained through week 24. At week 12, the proportion of patients who met or exceeded the MCID for placebo, baricitinib 2 mg and baricitinib 4 mg, respectively, was 40%, 57% and 53% (p=0.001 for baricitinib 2 mg vs placebo; p=0.006 for baricitinib 4 mg vs placebo) and was 34%, 56% and 56% at week 24 (p=0.001 for both baricitinib groups vs placebo). For the SF-36 MCS measure, numeric, but not statistically significant, differences in the change from baseline were found between the baricitinib-treated groups versus placebo. The proportion of patients who met or exceeded the MCID was not statistically significantly different from placebo for either baricitinib treatment group (figure 2B).
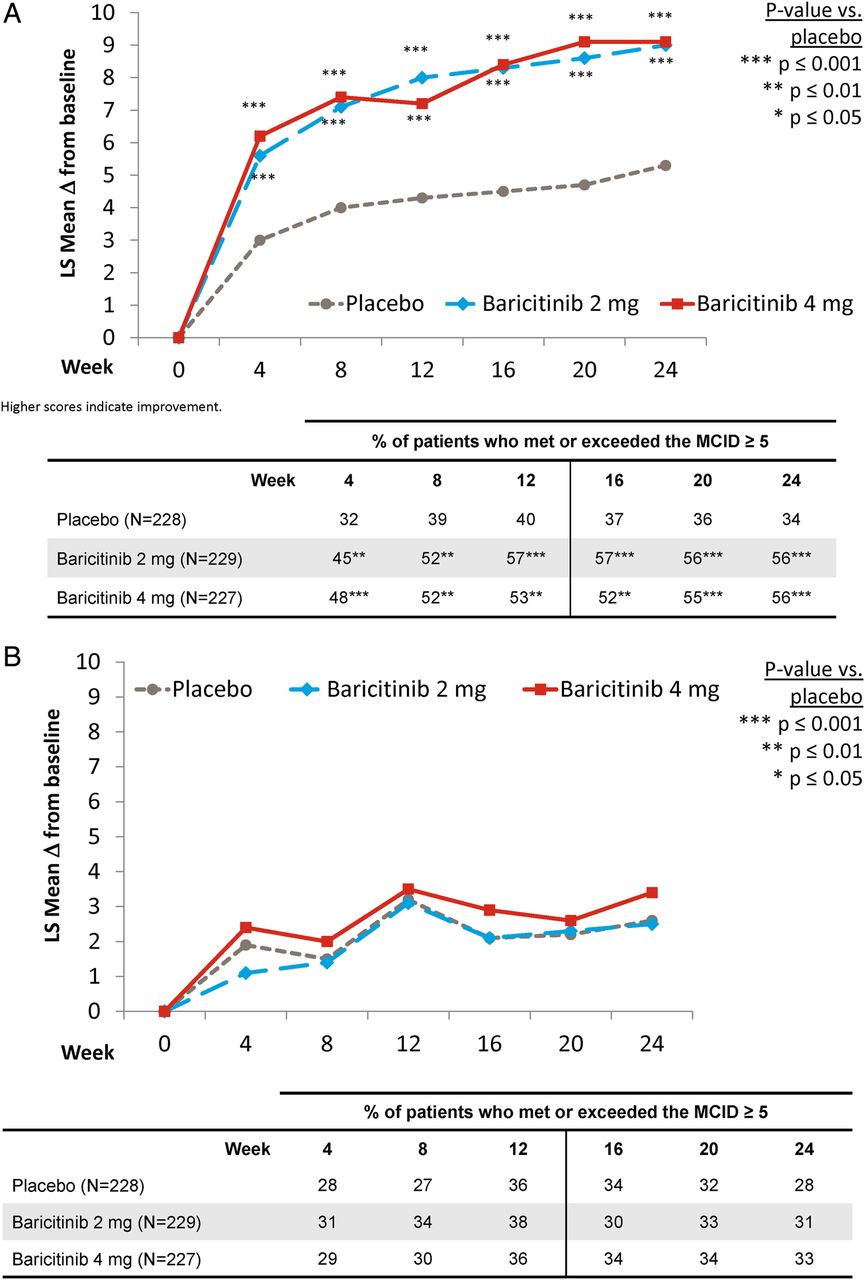
{kind=link}
{kind=link}
Change from baseline for the physical and mental component score for the SF-36. (A) Physical component score. (B) Mental component score.
EuroQol 5-Dimensions
At weeks 12 and 24, statistically significant improvement in the EQ-5D UK index score was observed for both baricitinib treatment groups versus placebo (table 3). A statistically significant improvement in the EQ-5D UK index score was observed at the first postbaseline assessment, week 4, for baricitinib 4 mg versus placebo, but not for baricitinib 2 mg versus placebo (data not shown).
Least-squares mean change from baseline at 12 and 24 weeks for patient-reported outcomes
For the EQ-5D VAS, at week 12, statistically significant improvement in the EQ-5D VAS was observed for both baricitinib treatment groups versus placebo (p=0.001 for both baricitinib groups vs placebo); this was maintained through week 24 for the 2 mg group (p=0.005 vs placebo) but not for the 4 mg group (p=0.159 vs placebo) (table 3). Baricitinib-treated patients were not significantly different from placebo-treated patients at week 4, the first data assessment (data not shown). Similar results were observed with the US algorithm for the EQ-5D (table 3).
Work productivity and activity impairment
At baseline, only 34–40% of the patients were employed (table 4). Patients treated with baricitinib 2 or 4 mg reported statistically significantly improved regular activity compared with placebo-treated patients at week 12 (p=0.004 for baricitinib 2 mg vs placebo and p=0.003 for baricitinib 4 mg vs placebo) but not at week 24 for either group (p=0.156 for baricitinib 2 mg vs placebo and p=0.179 for baricitinib 4 mg vs placebo; table 4). Among those patients who were employed at baseline and those who maintained employment at weeks 12 or 24, statistically significant improvements with respect to presenteeism for baricitinib 2 mg and work productivity loss for baricitinib 2 mg and 4 mg were seen compared with placebo at week 12 but not at week 24 (table 4). No statistically significant differences between groups were seen for absenteeism.
Work Productivity and Activity Impairment Questionnaire-Rheumatoid Arthritis (WPAI-RA) at baseline and at 12 and 24 weeks
Discussion
In this analysis of the Phase III RA-BUILD trial, baseline PROs revealed long duration (∼90 min) and severity of MJS, severe impairment of physical function and high levels of pain and fatigue (including tiredness) among patients with RA and an IR to csDMARDs, who had not previously been treated with a biological DMARD. Once-daily baricitinib 2 or 4 mg produced significant improvements compared with placebo in most of the prespecified PROs, including physical function, fatigue, duration and severity of MJS, pain and HRQOL. For some PROs, including selected daily diary measures, treatment with the 4 mg dose resulted in more rapid improvement compared with the 2 mg dose.
The PROs demonstrated a rapid onset of action for baricitinib; statistically significant improvements were seen as early as week 1 in the HAQ-DI, PtGA and the patient assessment of pain, and were maintained until the end of the trial at week 24. Similar results were seen with severity of MJS, Worst Tiredness, Worst Joint Pain as assessed using the patient diaries,18 and the PROs continued to improve to week 12. Consistent with these results, patients treated with baricitinib 2 or 4 mg reported greater improvement in HRQOL, as measured by the EQ-5D and SF-36 PCS, compared with placebo-treated patients. For the SF-36 component scores, a change from baseline of either 2.5 or 5 is typically considered an MCID.27 ,28 This analysis applied the higher threshold for the MCID and found that approximately half of the baricitinib-treated patients reported scores that met or exceeded the MCID for the SF-36 PCS (figure 2A). In contrast with the SF-36 PCS, no statistically significant differences were observed between baricitinib-treated and placebo-treated patients with the SF-36 MCS. Across treatment groups at baseline, the SF-36 MCS values ranged from 45 to 46, which are close to 50, the population norm.27 This suggests limited impairment for the SF-36 MCS at baseline; consequently, a marked improvement with therapy would not have been expected. The SF-36 MCS result aligns with previous results from other trials of baricitinib.34–36
The WPAI-RA data are difficult to interpret, as the number of patients who were employed at baseline and employed throughout the trial was small, and the 24-week duration is most likely a relevant limitation for this measure. A study with a longer duration and a focus on work productivity may provide more definitive evidence regarding the potential treatment effects of baricitinib on work productivity.
The results from this analysis are similar to those observed in three other phase III randomised clinical trials of baricitinib in different patient populations.34–36 Additionally, the improvements in the PROs with baricitinib in the current analysis were directionally consistent with the results reported with approved biological or targeted synthetic DMARD therapies in analogous patient populations.37–40
The limitations of this analysis include the use of carrying forward the last observations before rescue or discontinuation as a method of imputation for some continuous measures. Although an appropriate method of handling such data, this method assumes that the PRO values would not change over time if these events had not occurred, an unverifiable assumption. Most patients (89%), however, completed the trial;18 sensitivity analyses assessing missing data (data not shown) suggest that the study results would not be influenced by this data handling method. Additionally, this is a short-term study of 24 weeks, which does not allow for an assessment of the long-term treatment effects of baricitinib. This may particularly affect work productivity assessments. The employment status of patients in this study did not improve significantly, most likely because of the short duration of the trial; patients with disease-associated unemployment are unlikely to recover, then search for and find new work within 6 months. Furthermore, patients from 22 countries participated in the trial; the different rates of unemployment and policies related to workplace accommodations for RA may have influenced a patient's employment status.
This study has used a number of established PRO measures that assess outcomes of interest for patients. Some measures are incorporated in the ACR core set measures, and others, such as the EQ-5D and SF-36, are commonly used HRQOL instruments that may more broadly assess the effects of RA and its treatment on patients. Information from PROs may help the healthcare provider and patient determine the optimal treatment plan for the patient by (1) identifying the onset of action for when the patient will feel an improvement in symptoms, (2) evaluating the length of time the patient should try the treatment before determining that it is ineffective and (3) assessing if the treatment is still efficacious after initial improvement. Observing positive changes in PROs may help the patient adhere to therapy, an important element of treatment effectiveness. Additionally, the inclusion of PRO measures in clinical trials also helps to compare results across studies and patient populations and to assess the broader societal impact of RA and treatment for it.
In conclusion, the RA-BUILD study demonstrated that treatment with baricitinib 2 or 4 mg provided a significant improvement compared with placebo in most PROs across different domains of RA, including physical function, MJS, fatigue, pain and HRQOL. Improvements tended to occur rapidly, most notably for the 4 mg dose, and were maintained throughout the 24-week trial. These results confirm that the efficacy of baricitinib is not limited to physician-based or laboratory-based assessments, but that efficacy translates to a positive benefit on patients' quality of life and overall function. Consistent with prior observations, these data support a conclusion that baricitinib is a potentially valuable addition to the RA treatment arsenal for patients struggling with this common and disabling condition.
Acknowledgments
This study and manuscript were sponsored by Eli Lilly and Incyte Corporation. The authors would like to thank Molly Tomlin, MS, of Eli Lilly and Company for her assistance with manuscript preparation and process support and Scott Beattie, PhD, for statistical support.
References
Footnotes
Contributors All authors have made substantial contributions to the intellectual content of the manuscript. Specifically, these authors contributed to the acquisition of the study data: PE, RB, JMC, Y-CC, SdB, TR and MD. These authors participated in the conception of the study: PE, MD, SdB, TR, CLG and AMD. These authors analysed the data for the study: JL. All authors assisted in the interpretation of the data for the paper, provided critical revision of the paper and gave final approval for the paper's submission.
Funding Eli Lilly and Company and Incyte Corporation.
Competing interests PE has received grant/research support or consulting support from Abbott, AbbVie, Bristol Myers Squibb, Eli Lilly and Company, MSD, Novartis, Pfizer, Roche, Samsung, Takeda and UCB. RB has received research grants and/or participated in advisory boards from Abbvie, BMS, Janssen, Novartis, Pfizer, Lilly, MSD and Roche. JMC has received research grants and speaker fees from Abbott (AbbVie), Bristol Myers Squibb, Boehringer Ingelheim, Eli-Lilly, Merck Sharp Dohme, Novartis, Pfizer, Roche, Sanofi-Aventis, Schering-Plough and UCB. Y-CC has received speaker's bureau fees and/or grant research support from AbbVie, Bristol Myers Squibb, Eli Lilly and Company and Pfizer. CLG, AMD, SdB, JL, TR and CH-CC are full-time employees of Eli Lilly and Company and may own stock or stock options in Eli Lilly and Company. MD has received grant/research support or consulting support from AbbVie, Bristol Myers Squibb, Eli Lilly and Company, Novartis, Pfizer, Roche, Sanofi and UCB.
Ethics approval The Ethics Committee from each participating centre approved the study.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Additional data may be available on request from the study sponsors.