Article Text

Abstract
Osteocytes are the most abundant cell type in bone and are distributed throughout the mineralised bone matrix forming an interconnected network that ideally positions them to sense and to respond to local biomechanical and systemic stimuli to regulate bone remodelling and adaptation. The adaptive process is dependent on the coordinated activity of osteoclasts and osteoblasts that form a so called bone multicellular unit that remodels cortical and trabecular bone through a process of osteoclast-mediated bone resorption, followed by a phase of bone formation mediated by osteoblasts. Osteocytes mediate their effects on bone remodelling via both cell–cell interactions with osteoclasts and osteoblasts, but also via signaling through the release of soluble mediators. The remodelling process provides a mechanism for adapting the skeleton to local biomechanical factors and systemic hormonal influences and for replacing bone that has undergone damage from repetitive mechanical loading.
- Osteoporosis
- Osteoarthritis
- Bone Mineral Density
- Cytokines
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Throughout postnatal life bone undergoes continuous structural reorganisation and adaptation. The adaptive process is dependent on the coordinated activity of osteoclasts and osteoblasts that form a so called bone multicellular unit (BMU) that remodels cortical and trabecular bone through a process of osteoclast-mediated bone resorption, followed by a phase of bone formation mediated by osteoblasts.1 The remodelling process provides a mechanism for adapting the skeleton to local biomechanical factors and systemic hormonal influences and for replacing bone that has undergone damage from repetitive mechanical loading.2 ,3
Importantly, under physiological conditions, the amount of bone that is removed during the phase of bone resorption is matched by the amount of bone that is added by bone formation. The mechanisms involved in ‘coupling’ of bone resorption and formation are mediated in part by the release of bone growth factors, including transforming growth factor-β (TGF-β) and bone morphogenetic proteins (BMPs), which are released from the bone matrix during the phase of bone resorption.4 ,5 Osteoclasts provide an additional source of coupling factors including BMP-6, sphingosine-1-phosphate and Wnt-10b that induce osteoblast-mediated bone formation.6 ,7 Osteoclasts also produce factors that inhibit osteoblast differentiation and activity.8
Although there have been major advances in identifying the mechanisms involved in the regulation of osteoclast and osteoblast differentiation and activity, the specific cellular events associated with activation of BMUs and the initiation of bone remodelling until recently have been a matter of controversy. Rodan and Martin9 first proposed a key role of osteoblasts in initiating bone remodelling through the release of pro-osteoclastogenic mediators. Recent studies using a variety of experimental approaches have, however, challenged this concept. For example, Corral et al10 and more recently Galli et al11 used genetic approaches to delete mature osteoblasts in mouse models and showed that this did not affect osteoclast-mediated bone resorption. Importantly, morphological studies demonstrate that mature bone forming osteoblasts are not present during the activation of the BMU,1 indicating that alternate mechanisms must be involved in the initiation of bone remodelling.
Several lines of evidence have now established a key for osteocytes in the regulation of bone remodelling.12 ,13 Osteocytes are the most abundant cell type in bone and are distributed throughout the mineralised bone matrix forming an interconnected network that ideally positions them to sense and to respond to local and systemic stimuli to regulate bone remodelling and adaptation. These effects are mediated via both cell–cell interactions with osteoclasts and osteoblasts but also via signalling through the release of soluble mediators. Recent studies by Xiong et al14 and Nakashima et al15 provide convincing evidence supporting a critical role for osteocytes in regulating the effects of mechanical loading on bone remodelling. They deleted receptor activator of NF-κB ligand (RANKL), the master regulator of osteoclastogenesis, in osteocytes and showed that the animals developed an osteopetrotic phenotype and were resistant to bone loss induced by mechanical unloading using tail vein suspension. These findings support the observations of Tatsumi et al16 who used genetic approaches to selectively ablate osteocytes and showed that the animals lost the capacity to increase bone loss in response to unloading. Osteocytes also are a source of osteoprotegerin (OPG), the potent inhibitor of RANKL,17 and differential production of OPG and RANKL by osteocytes provides a unique mechanism by which osteocytes can regulate osteoclast-mediated bone resorption (figure 1).

Osteocyte-derived receptor activator of NF-κB ligand (RANKL) increases bone resorption. Osteocytes control unloading-induced bone loss by induction of RANKL in osteocytes, which results in increased osteoclast-mediated bone resorption.
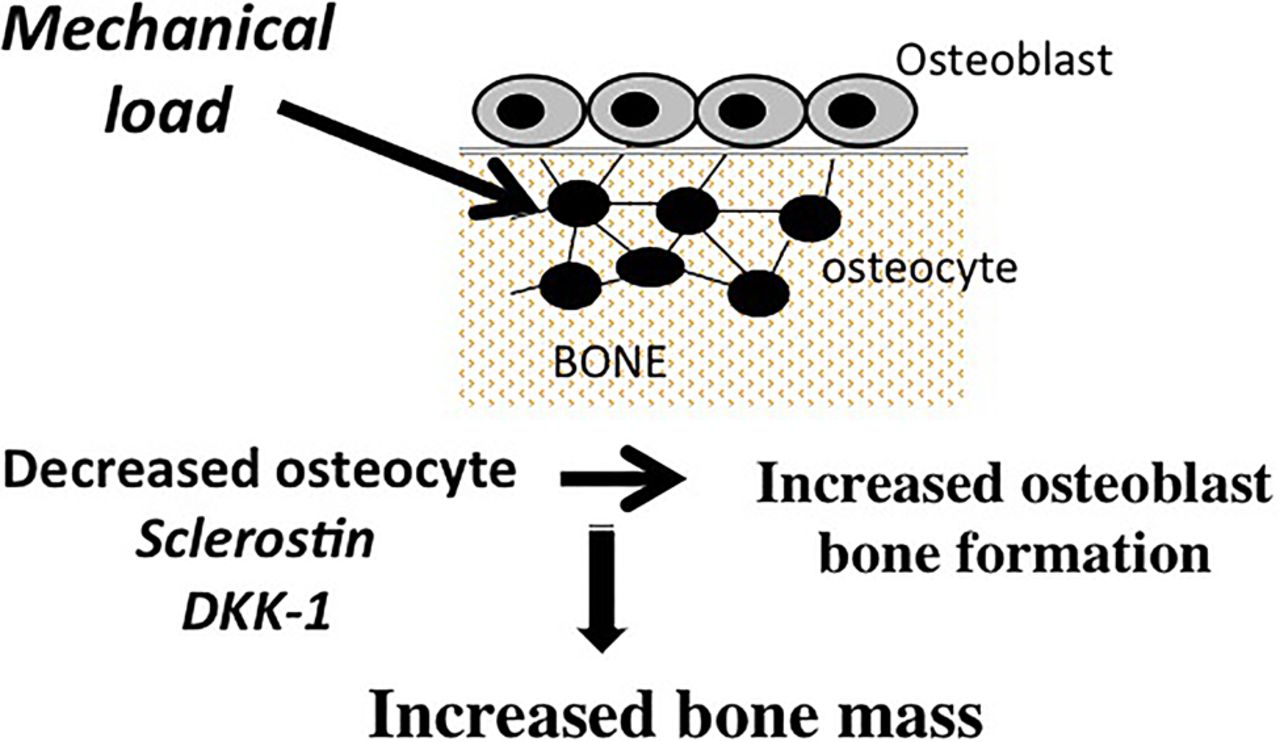
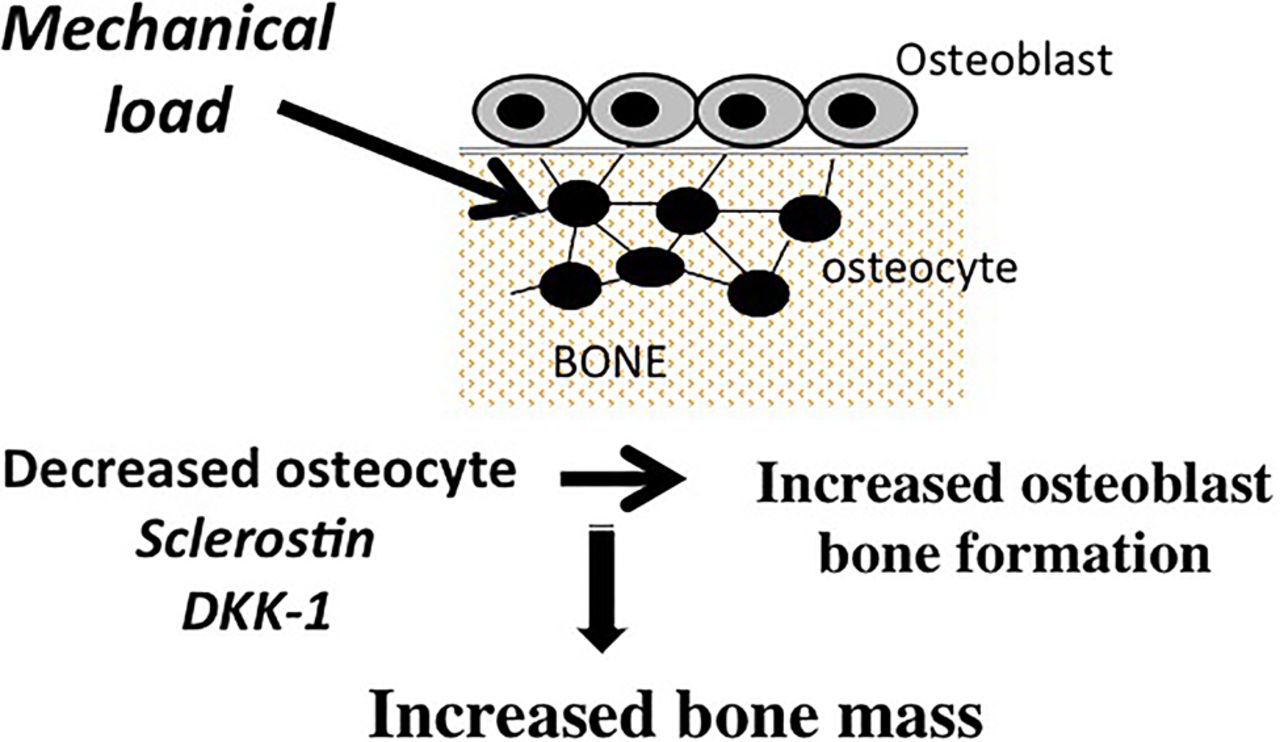
In addition to OPG and RANKL, osteocytes are the source of a diverse array of products that have the capacity to modulate bone remodelling, including small molecule mediators such as prostanoids, nitric oxide, and nucleotides, as well as a broad spectrum of cytokines and growth factors such as insulin-like growth factor-1 (IGF-1), vascular endothelial cell growth factor (VEGF) and TGF-β.13 ,18–24 Osteocytes also are a major source of fibroblast growth factor 23, which regulates serum phosphorus levels by increasing renal phosphate excretion.25 Recent studies have drawn attention to the role of two osteocyte-derived products, sclerostin and Dickkopf-related protein 1 (DKK-1), in the regulation of bone remodelling. These molecules are potent and specific inhibitors of the Wnt/β-catenin pathway, which plays a major role in the regulation of osteoblast-mediated bone formation.26 Robling and co-workers were the first to show that mechanical loading decreased the expression of sclerostin and DKK-1 in osteocytes. The decrease in the production of these Wnt pathway inhibitors by osteocytes resulted in upregulation of β-catenin signaling in osteoblasts and an associated increase in bone formation27–29 (figure 2). In contrast, unloading resulted in increased expression of sclerostin and DKK-1 and reduction in bone formation. Osteocytes also play a role in mediating the effects of parathyroid hormone (PTH) on bone formation. These effects are in part attributable to PTH-induced suppression of sclerostin by osteocytes.30–32 In addition, there is evidence that PTH-induces increases in RANKL production by osteocytes, indicating a key role of osteocytes in mediating the effects of PTH on osteoclast-mediated bone resorption.33
{kind=link}
{kind=link}
Osteocyte-derived sclerostin and Dickkopf-related protein 1 (DKK-1) regulate bone formation. Loading decreases osteocyte-derived sclerostin and DKK-1, which results in activation of the Wnt/β-catenin signaling in osteoblasts and increased bone formation.
Studies by Morse et al34 have provided further insights into the role of osteocyte-derived sclerostin in regulation of bone formation. They examined the effects of loading and unloading in mice in which the Sost gene was deleted in osteocytes. Sclerostin is the product of the Sost gene and deletion of Sost resulted in loss of sclerostin production by osteocytes. They found that mice lacking the gene maintained the capacity to increase bone mass in response to loading, indicating the existence of sclerostin-independent mechanisms for regulating bone formation. In contrast, bone loss was attenuated in the mice that lacked the Sost gene, indicating that osteocyte-derived sclerostin played a key role in mediating the effects of unloading on bone formation. The loss of bone was attributable to a decrease in bone formation but also an effect on bone resorption, indicating that in addition to effects on bone formation, sclerostin and the Wnt/β-catenin pathway in osteocytes also contributes to the regulation of osteoclast-mediated bone resorption. This effects is in part related to a decrease in osteocyte-derived OPG resulting in a shift in the RANKL/OPG ratio favouring increased RANKL-mediated osteoclastogenesis.
As discussed above, in addition to the role of bone remodelling in providing a mechanism for adapting the skeleton to mechanical loading and systemic hormones, BMU-mediated bone remodelling also provides a mechanism for replacing bone that has undergone damage from repetitive mechanical loading. The term ‘targeted remodelling’ has been used to identify the process of remodelling of bone that has undergone microdamage.2 ,3 Studies by Schaffler et al have provided insights into the mechanisms of targeted remodelling.13 ,21 ,35 ,36 They showed that the remodelling process at sites of microdamage is initiated by apoptosis of osteocytes at the sites of bone damage, and that osteocytes adjacent to the dying cells release products, including RANKL, VEGF, ATP, sphingosine-1-phosphate and chemokines that activate endothelial cells and recruit bone cell precursors, including osteoclasts and osteoblasts, to the site of injury where they can repair the damage through BMU-mediated remodelling. Targeted remodelling of subchondral bone at sites of excessive mechanical loading play a contributory role in the adaptive bone changes in osteoarthritis (OA).
Several recent reports have provided insights into the potential contributory role of osteocyte-derived sclerostin and the Wnt/βcatenin pathway in the development of OA bone pathology.37–40 For example, Chan et al38 examined the expression of sclerostin in osteochondral samples from human subjects with OA and sheep and mice with surgically induced OA. They found decreased expression of sclerostin in osteocytes in regions corresponding to sites of increased mechanical loading and associated increased bone formation. Appel et al37 also observed evidence of decreased sclerostin expression in regions of subchondral bone pathology in samples from patients with OA. In other studies Funck-Brentano et al39 used the TOPGAL reporter mice to assess activation of Wnt/β-catenin signaling in joint tissues. This system permits detection of the specific cellular localisation of activation of the Wnt/β-catenin signaling pathway using X-gal staining of the tissue sections. They found that X-gal staining was increased in osteocytes in subchondral bone at sites of increased subchondral bone formation. They also showed increased VEGF in osteoblasts and osteocytes in the subchondral bone in menisectomised mice with OA.
In summary, recent studies have identified osteocytes as the key regulators of bone remodelling in both physiological as well as pathological states. Osteocytes are widely distributed throughout the bone matrix and are optimally positioned to sense and respond to changes in mechanical loading and to local bone damage. They also have the capacity to regulate bone remodelling in response to systemic hormones. Osteocytes exert their effects on bone remodelling via direct cell–cell contacts and by the release of soluble mediators that control the recruitment, differentiation and activity of osteoclasts and osteoblasts. Targeting osteocytes and their products represents a novel approach to treating skeletal disorders associated with de-regulated bone remodelling.
References
Footnotes
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.
Data sharing statement No additional data are available.