Article Text

Abstract
Rheumatoid arthritis (RA) is a systemic autoimmune disease characterised by the presence of autoantibodies. Their value for diagnostic/prognostic purposes has been well established. In contrast, their role in established disease and their associations with disease activity is less clear. Moreover, as evidence is accumulating that these autoantibodies are causally involved in certain key aspects of the disease, such as the initiation and perpetuation of joint inflammation or join destruction, autoantibodies in RA can no longer be regarded as mere epiphenomena, but are integral elements of the pathophysiology of RA.
- Rheumatoid Arthritis
- Rheumatoid Factor
- Autoantibodies
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
Rheumatoid factor and antibodies against citrullinated peptides/proteins (ACPA) are directly involved in the inflammatory response in rheumatoid arthritis.
Autoantibodies are associated with disease activity and with joint damage.
Levels of autoantibodies change during disease/on treatment.
Introduction
Rheumatoid arthritis (RA) is a complex chronic disease. Historically, RA has been labelled as seropositive or seronegative, according to the presence or absence of detectable rheumatoid factor (RF). RF is an antibody directed against the Fc portion of human IgG. It is well characterised, although its exact origin still remains unclear. Typically, RF is of IgM isotype, but IgG and IgA may also occur. In the past, RF levels were determined by classical agglutination reactions; however, sensitivities and specificities depended on the type of test (eg, latex fixation test, or Waaler-Rose test using sheep erythrocytes). Recently, RF levels are determined by nephelometry.1
Other antibodies with specificities for RA include antiperinuclear factor and antikeratin antibodies which were known for a long time, but have never reached widespread use because of difficulties in their detection.2 ,3 Recently, a large portion of patients with RA were shown to produce antibodies recognising certain peptides containing citrullinated arginine residues. The autoantibodies that were detectable in this way were, therefore, summarised as anti citrullinated peptide/protein antibodies (ACPA) and this is their current name. When cyclic citrullinated peptides (CCP) were used instead of linear citrulline as antigens in test systems, diagnostic test properties further improved and these test were then commercialised as anti-CCP tests.4 Suddenly, patients could also be classified as seropositive or seronegative for RFs, as well as for ACPA. Today, second and third generation of anti-CCP assays are available and have expanded the number of antigens to further improve sensitivity.5 Recent evidence suggests that these two groups of autoantibodies might not be the only ones present in RA. Anticarbamylated protein antibodies are present particularly in patients with RA in a significant amount and are prognostically seem to be similar to ACPAs. Although these do coincide with both ACPA and RF to a large extent, there is a fraction of about 10% of ACPA-negative RA patients who test positive, thus closing the seronegative gap of RA a bit more. Whether this has implications for diagnosis or subsequent treatment decisions remains to be seen.6 ,7
Purpose of the review
With all these established and novel serological characteristics and tests at hand, the purpose of the present review is to assess the management implications of autoantibodies in RA. Do these tests affect or change management of RA in any respect and what are the potential scientific explanations for this?
Our review will span the diagnostic associations of these autoantibodies, their link to disease activity in established RA, and their prognostic associations. We will discuss the potential pathogenetic mechanisms that may underlie these observable associations. Some of these clinical associations are important clues to direct pathogenetic hypotheses. Finally, epidemiological data suggest that serological markers, particularly RF, are not fixed features of a patient's disease, but may exhibit change through the course of RA, at least in some patients. We will discuss this at the end of the review.
Diagnostic associations of autoantibodies for RA
RF and ACPA have been associated with the diagnosis of RA. RF has been associated with a diagnosis of RA when used in patients with early undifferentiated arthritis in multiple studies.8–12 According to one review of the topic, the positive likelihood ratios of RF for a diagnosis of RA ranged from 1.1 to 13.5.13 The presence of ACPA has likewise been shown to predict later development of RA in many studies of early arthritis.4 ,14–17 Both antibodies have been shown to predict RA even when detected in healthy individuals.18–20 The clear diagnostic value of these tests is emphasised in the prominent position of serology in management recommendations for early arthritis, as well as in current classification criteria for RA.21 ,22 Another older review of the diagnostic properties of RF and ACPA suggested higher specificity for ACPA;23 however, publication bias for the then limited number of ACPA studies could not be ruled out. RF may to a greater extent than ACPA be present in healthy, elderly persons or in patients with other autoimmune or infectious diseases.24 However, it has been suggested that raising the standard cut-point of positivity for RF to 50 U/mL increases the specificity of RF for a diagnosis of RA to at least match the one for ACPA.14
Given all this epidemiological evidence, it is worthwhile to discuss the role of RF and ACPA in the pathogenesis of RA. The development of ACPA has strongly been linked to the presence of the shared epitope, a sequence motif of HLA-DRB1 that can be found in many patients with RA, especially in smokers. This combination of genetic and environmental risk factors reflects the complex pathogenesis of systemic autoimmune diseases, such as RA, and demonstrates that many factors contribute to disease development. Therefore, the mere presence of either ACPA or RF is not sufficient to cause disease. However, analysis of ACPA specificities and titres in longitudinal analyses of ACPA-positive individuals for over 10 years before development of symptoms showed a dramatic increase in titre of various ACPA 6 months before the onset of RA, suggesting events leading to precipitation of the disease also increase ACPA titres.25 Similar observations have been made for anticarbamylated antibodies that also develop well before the onset of symptoms and exhibit similar kinetics like anti-CCP antibodies. However, these are not associated with the classical risk factors for ACPA, such as HLA-DR4 and smoking, suggesting different pathophysiological processes in their generation.26 It is interesting to note that RF appears much later (ie, much closer to disease onset) as compared with ACPA.27 Also its presence in combination with ACPA strongly increases the chances of developing RA in patients with arthralgia,28 which could suggest that RF is needed to trigger overt disease in ACPA-positive individuals. This supports previous notions of a potential increasing risk for developing RA with increasing titres of RF.29
Role of autoantibodies in the inflammatory disease process
There is a strong scientific basis to support the contributions of RF to the inflammatory disease process. First, RF has been shown, by complexing ACPA, to induce proinflammatory cytokines like tumour necrosis factor (TNF), interleukin 1β, IL-6 and IL-8 (figure 1A).30 Synovial fluids of RF-positive RA patients have been observed to contain immune complexes and exhibit low complement levels,31–33 suggesting activation of complement by immune complexes. It is also well established that immune complexes of RA joints contain RF.34 ,35 Immune complexes, by binding to Fc receptors, especially Fcγ receptors on human monocytes ex vivo, have been implicated in mediating the proinflammatory effects,36 ,37 particularly by subsequently activating the production of TNF (figure 1A). The latter induces a cascade of other cytokines, molecules and cells,38 and its inhibition is a key therapeutic strategy in RA. It has to be noted, however, that the biology of Fc receptors and Fcγ receptors, in particular, differs between mice and men; therefore the exact roles, especially of Fcγ receptors, are still not fully understood. Nevertheless, an important involvement of activating Fcγ receptors is apparent in the pathogenesis of inflammatory arthritis in murine models as well as in the pathogenesis of RA that is also underlined by the fact that Fcγ receptor IIIA polymorphisms are risk factors for ACPA-positive RA.37 ,39 ,40 ACPA, particularly RF, has also been shown to increase NETosis from neutrophil granulocytes. NETs can activate synovial fibroblasts; however, they also contain citrullinated autoantigens, further fuelling ACPA production.41
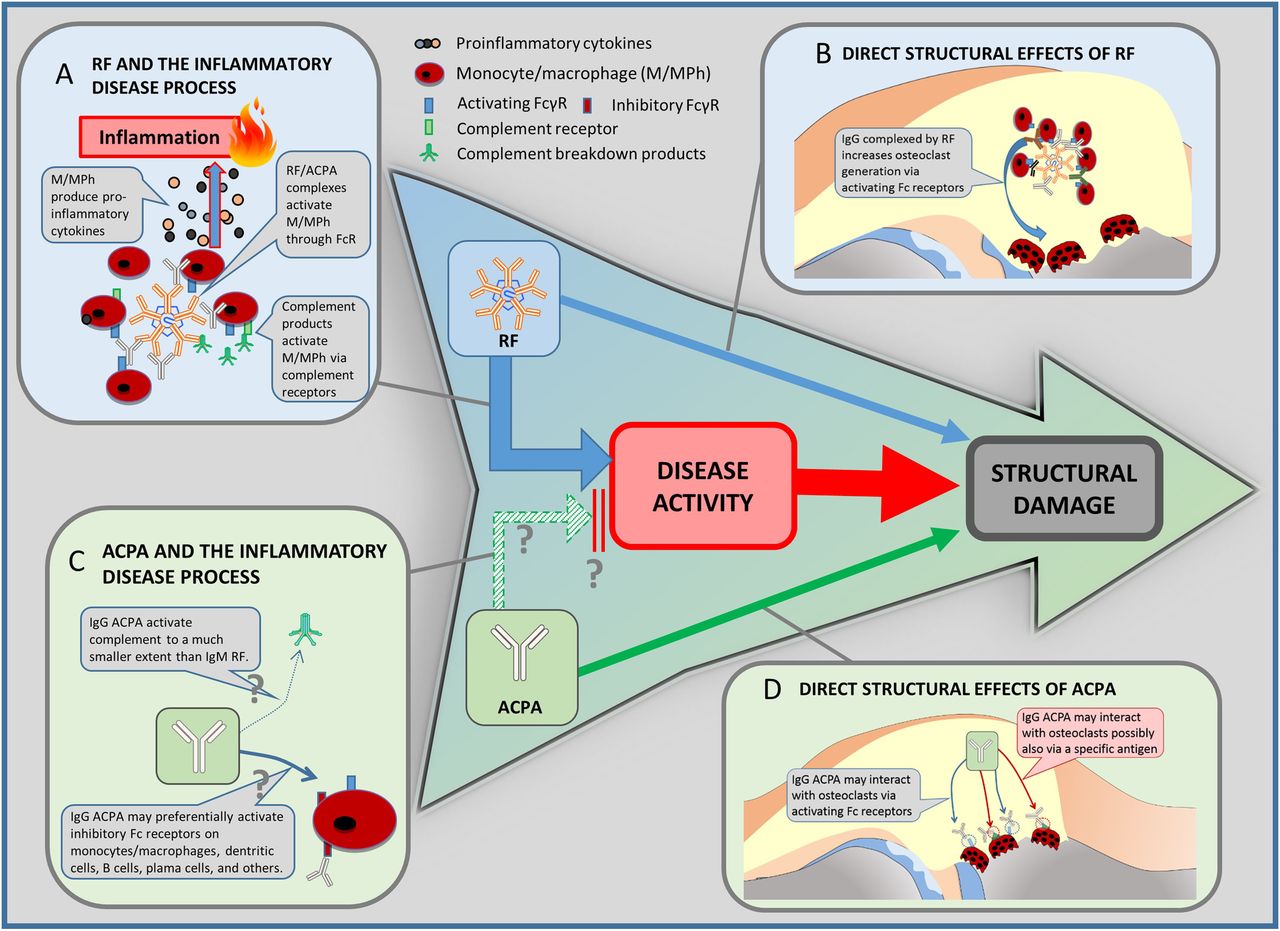
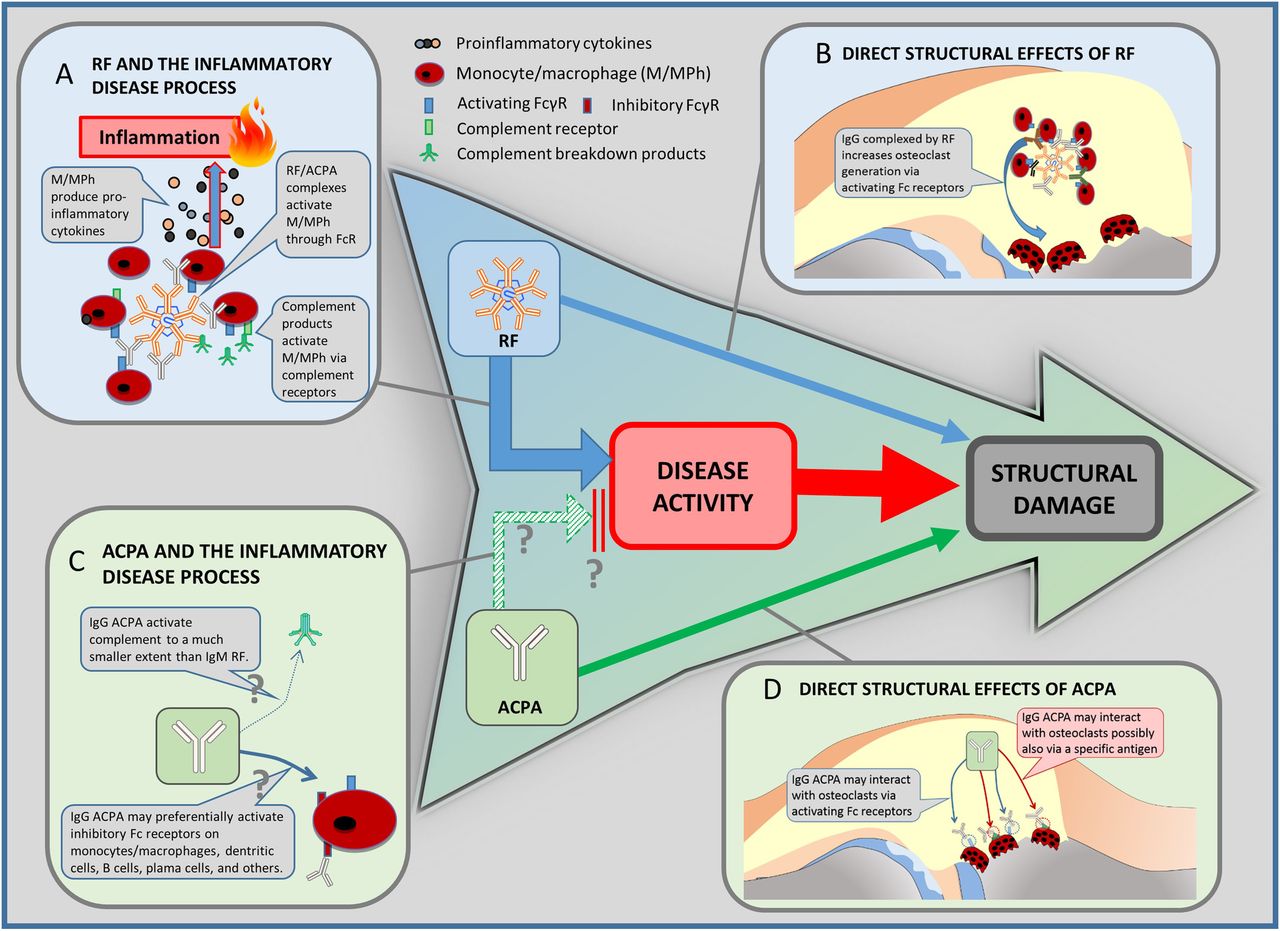{kind=link}
Clinical paradigms and pathogenetic explanations. The well-established clinical sequence of disease activity and structural damage is in the centre of the figure. At the same time, in clinical studies, rheumatoid factor (RF) has been shown to be linked to damage of rheumatoid arthritis via increasing disease activity, but also directly (blue arrows). Antibodies against citrullinated peptides/proteins (ACPA) is also associated with structural outcomes (solid green arrow). However, ACPA have not been shown to be associated with clinical disease activity. Vice versa, disease activity might even be lower in ACPA-positive patients (broken green arrow; see also, section ‘ACPA paradox’). Since this finding needs further confirmation, it is marked with a ‘?’. The panels around the central sequence are pathogenetic explanations for these clinical links; these links are quite well established for RF and the inflammatory disease processes (A), RF and direct effects on progression (B), as well as ACPA and its direct links to progression (D). They are hypothetical for the (paradoxic) link between APCA and disease activity (C). For details on the links and references see respective sections in the text.
In addition, some aspects relevant to the role of autoantibodies can also be studied in animal models, some of which heavily depend on the presence of autoantibodies.42 In collagen-induced murine arthritis models (CIA), the generation of anticollagen type II-specific antibodies is a condition sine qua non for the development of clinical arthritis. However, similar to the human situation, the presence of anticollagen antibodies does not suffice to induce arthritis, evidenced by the fact that there are several reports showing complete protection from the disease despite the presence of typical autoantibody levels.43–45 It is interesting to note that while in CIA anticarbamylated antibodies do develop, ACPAs have not been observed in this model; the relevance of these autoantibodies, however, has not been clarified as yet.46 This shows that even in animal models, where autoantigens as well as the development of autoantibodies can be investigated much more thoroughly as compared with humans, it is not yet completely understood how autoantibodies contribute to overt disease development. However, it was shown that modifications of the Fc portion of (auto)antibodies regulates their pathogenicity in an animal model that entirely depends on passively administered antibodies, as well as modulated osteoclast generation mediated by immune complexes.47 ,48 In humans, Fc modifications of ACPAs have been described. Consistent with the observations from the mouse models, these modifications modulate effector functions of ACPA, such as binding affinity to certain Fc receptors, and thus also help regulate the generation of osteoclasts.49 It has also been demonstrated that Fc modifications in patients undergo significant changes when ACPA-positive individuals progress to overt RA.50 The fact that antibody effector functions can be regulated by Fc modifications represents a new layer of complexity in the pathogenesis of autoantibody-associated diseases such as seropositive RA.
Associations of autoantibodies with clinical disease activity, and the ‘ACPA paradox’
In a cross-sectional perspective at the baseline of clinical trials, ACPA-positive patients present with less disease activity than ACPA-negative patients.51 This holds true in the subsets of RF-positive and RF-negative patients. Given the evidence about the strong association of ACPA with structural damage, these data may also speak for direct effects of ACPA on bone damage independent of disease activity. In fact, the possibility has been suggested that aside from Fc-mediated effects of ACPA on osteoclasts, certain ACPA might also recognise structures on osteoclasts directly, thereby promoting osteoclast generation (figure 1D).49 The important role of RF, and not of ACPA, in relation with the higher levels of disease activity was also implied in another recent study in which the authors have revealed that RF-positive patients regardless of ACPA status compared with RF-negative/ACPA-positive patients exhibited higher disease activity, as well as higher levels of proinflammatory cytokines.52 However, in that study it was also hypothesised that ACPA have a synergistic effect in the presence of RF regarding disease activity. These findings, therefore, support concepts that RF, and not ACPA, is the main serological inflammatory player while ACPA individually may not only not fuel RA disease activity, but may even mitigate it.
What are potential explanations for the clear relationship of RF with disease activity, as well as for its absence for ACPA (figure 1A, C)? First, as assessed in routine practice and clinical trials, RF is of IgM isotype while ACPA are mostly IgG antibodies; IgM isotype activates complement to a much larger extent than IgG,53 thus potentially leading to a stronger secondary inflammatory response via inflammatory complement breakdown products and/or complement receptor-mediated macrophage activation.54–56 In this setting, ACPA might preferentially activate inhibitory Fcγ receptors and thus mitigate the inflammatory response (figure 1C).57 At the same time, however, it is conceivable that ACPA IgM also exist, while current tests are only able to detect IgG.
Second, the B cells producing the autoantibodies are different. Most likely, ACPAs are produced by plasmablasts/plasma cells residing in the bone marrow or the inflamed synovial membrane.58 However, ACPA producing B cells have recently been identified as class-switched memory B cells and plasmablast by phenotypic analyses, and are most likely maturing in germinal centres, at least those that can be isolated from blood.59 It was also noted that the frequency of those peripheral ACPA-specific B cells correlated with ACPA titres, suggesting that these cells contribute to the overall amount of ACPA.59 In contrast, the source of RF is most likely a CD5+ subset of B cells. These CD5+ B cells might represent a particular subset of B lymphocytes, B1 B cells. The mentioned peripheral ACPA-producing plasma cells may have less plasticity and responsiveness to variations of the disease process, or might be less involved in cell–cell interactions in RA than the RF-producing B1 cells.60 ,61
B1 B cells are different from conventional B cells in several ways. First, these are able to produce their IgM antibodies independent of antigenic stimulation, that is, ‘blindly’ to several self-structures. The antibodies of B1 B cells usually are of low affinity and broadly reactive, and mostly directed against conserved structures, such as Fc portions of immunoglobulins or phosphorylcholine, as well as to viral antigens such as influenza. B1 B cells have been implicated in the clearance of apoptotic cells by providing antibodies recognising them, facilitating their non-immunogenic removal and thereby, contributing to immune homeostasis.62 These do not seem to require T cell help and no germinal centre reaction for producing their antibodies, which distinguishes them from the ACPA-producing conventional B cells. It should be noted that the existence of B1 cells in man has been a matter of some debates, although evidence of their existence is increasing63–65 and they have been related especially to RF production in humans long ago.66 However, evidence of plasticity of RF levels may support their existence, which would be one potential explanation of why changeability in levels is much smaller for ACPA (see last section ‘PLASTICITY OF AUTOANTIBODIES IN RA’). Also, the lack of association of APCA with disease activity needs to be clearly seen as distinct from the well-established association of ACPA with radiographic damage (see next sections ‘ASSOCIATION OF AUTOANTIBODIES WITH STRUCTURAL DETERIORATION’ and ‘PLASTICITY OF AUTOANTIBODIES IN RA’), which may still be well explainable through its direct effects on osteoclastogenesis.
Association of autoantibodies with structural deterioration
Similar to its diagnostic properties, the prognostic value of RF is also evident from a number of studies.4 ,14 ,67–69 The term ‘prognostic’ in these studies relates to structural outcomes, or erosive disease in particular. For ACPA, there are studies supporting the prognostic value regarding structural outcomes4 ,14 ,70 and functions.71 In one study, RF and ACPA were associated with premature death.72 Given these prognostic implications, consideration of presence or absence of autoantibodies has become part of contemporary management algorithms for RA, such as the treat-to-target strategy,73 or the management recommendations put forward by the European League Against Rheumatism (EULAR).74
However, these management strategies still mainly focus on disease activity—not on serology—given the paradigmatic belief that disease activity links into and causes structural progression over time in RA.75 ,76 A halt of the disease process, therefore, is believed to prevent bad disease outcome. Thus, the presence of chronic inflammation over time can be considered a prognostic outcome of RA; in fact, several additional prognostic studies link RF and ACPA to ‘persistence of synovitis’ or ‘treatment adaptation’ as outcomes in the prognostic setting, and have been reviewed previously.13 The association of ACPA and progression of structural joint damage has been demonstrated; the presence of anticarbamylated antibodies has been shown to be associated with increased radiographic damage.7 ,77 In addition, ACPA positivity has been shown to be associated with serum levels of receptor activator of nuclear factor κB ligand (RANKL), the key cytokine for osteoclast generation. Although not specifically analysed, this study also suggested additive effects of ACPA and RF on serum levels of RANKL.78
However, this paradigmatic link of RF with structural progression may not be as simple, and it is not clear whether it is mediated only via higher levels of disease activity or if seropositive disease in itself confers an independent risk for progression. In fact, in a recent study we found that the association between RF and structural progression in RA is primarily mediated by higher disease activity in RF-positive compared with RF-negative patients (figure 1, centre).79 However, in addition to increasing disease activity, the presence of RF conveyed a disease activity independent of the effect on joint damage, which was primarily related to bone erosions. Respective epidemiological data on ACPA currently do not exist.
Explaining the effects of RF that are unrelated to an increase of disease activity may be more difficult. However, RF by its ability to bind to any IgG is able to form large immune complexes. Interestingly, in steady state conditions, immune complexes seem to even inhibit osteoclast generation.80 ,81 However, under inflammatory conditions (as in the arthritic joint), at least in mice murine models, upregulation of activating and downregulation of inhibitory Fc receptors directs the response of OC precursors towards increased osteoclast generation (figure 1B).81 In addition, at least in experimental arthritis, osteoclastogenesis of immune complexes increases due to increased abundance of IgG subtypes that bind activating Fc receptors.81 On binding to Fcγ receptors on macrophages activate via the common γ chain, the spleen tyrosine kinase (Syk) signal transduction cascade.82 ,83 Of the Syk signals, via at least two pathways, one leads to induction of mitogen-activated protein kinases (MAPK), especially c-Jun N-terminal kinases (JNK), which induce proinflammatory cytokines in line with the above notion on the increase of disease activity; these cytokines, however, can also activate RANKL, a pivotal osteoclastogenic molecule. Moreover, Syk also induces nuclear factor of activated T cells c1 (NFATc1) via phospholipase Cγ (PLCγ). NFATc1 is yet another important transcription factor for osteoclast generation.84 In fact, this explanation would also reflect the clinical observation that activity independent effects were significantly associated only with progression of joint erosion, but less (and not significantly so) with cartilage damage.79 The fact that clinical trials of Syk inhibitors have provided only limited clinical benefit may not necessarily speak against this explanation, as the main driver of disease activity are the proinflammatory cytokines. The structural effects of Syk inhibition will be likely to remain elusive, as the clinical studies were stopped in phase II of the development programme.
The therapeutic implications of the epidemiological findings is that RF is prognostically important on several independent streams, and its consequences to structure may only be partly taken care of by treating RA disease activity. This is fully in line with the current management recommendations by the EULAR,74 which have lifted RF up to represent an independent bad prognostic factor: in patients with an insufficient response to methotrexate, RF status guides the ensuing treatment strategy. In other words, seropositive RA is considered to require more intensive therapy in patients newly started on methotrexate than seronegative RA at the same level of disease activity; treatment targets and strategies in RA may thus differ in seropositive and seronegative patients. They may—and probably should—not solely be based on a specific disease activity level.
Plasticity of autoantibodies in RA
It is interesting to note that epidemiological data suggest that serological markers, particularly RF, are not a fixed feature of a patient's disease, and these may change through the course of the disease, at least in some patients.85–88 This change in autoantibody levels has been reported to be more prevalent, and to occur in a greater extent in patients who also respond clinically.85 ,87 ,88 Seroconversions may indicate a profound response of RA, particularly as conversions occur more frequently in patients who respond well clinically. It is, however, unclear why some patients ‘respond’ with their serological markers and even ‘seroconvert’ to a negative status, while others do not, even if they respond clinically.
The change in RF is generally reported to be much greater than the change in ACPA.85 ,89 ,90 However, this might be a consequence of the different isotypes of these two autoantibodies (AABs), with RF mostly being measured as IgM class, whereas ACPA, as measured by ELISA, are of the IgG class, which may show less rapid responsiveness to therapeutic interventions. As discussed above, possibly different cell types are involved in the production of RF and ACPA, which may lead to the observed differences in plasticity of serological levels of these autoantibodies (see previous section ‘ASSOCIATIONS OF AUTOANTIBODIES WITH CLINICAL DISEASE ACTIVITY, AND THE ‘ACPA PARADOX’’). This is also highlighted by the observation that reduction of serum levels of RF is more pronounced than the reduction of ACPA on treatment with rituximab. Interestingly, however, ACPA levels seem to be more prone to change on treatment with rituximab than with other therapeutic agents, which also suggests that ACPAs too are not entirely produced by long-lived plasma cells, which are not targeted by RTX.91 ,92 Also, in simple correlation analyses performed as part of one study,85 changes in acute phase reactants (C reactive protein (CRP) and erythrocyte sedimentation rate (ESR)) were similarly associated with changes in ACPA as with changes in RF, while changes of swollen joint count (SJC), pain and patient global assessment (PGA) were only correlated with changes in RF. The latter finding is very intriguing and one can only speculate that RF may be more strongly involved in activating cytokine production within the joints, and that these local events are linked with swelling and pain. Indeed, pain has been suggested to be associated with enhanced release of proinflammatory cytokines, such as IL-6.29 IL-6 is also involved in B cell differentiation and the development of antibody-producing plasma cells.30 This might constitute a link between all these variables.
Conclusions
Although the detailed pathogenetic events are not completely resolved, the assessment and measurement of autoantibodies in RA is clearly relevant. There is an evident association of RF with the diagnosis of RA. Its prognostic link to structural progression is mediated mainly through an increase of disease activity, as well as putatively independently of disease activity via direct effects on osteoclastogenesis. There is likewise a clear link of ACPA with diagnosis of RA, and its structural progression. The effect of ACPA on disease activity, however, is paradoxical as ACPA-positive patients seem to present with less clinical activity than ACPA-negative patients. Although some explanatory hypothesis may be put forward, this finding urges further clinical and pathogenetic investigations. Finally, both levels of RF and ACPA have been reported to be changeable, particularly in patients who also improve their disease activity during therapy. This effect is stronger for RF than for ACPA.
All this ultimately challenges the concept of seropositivity as a disease characteristic, and may indicate the role of RF and ACPA for diagnostic purposes, as well as in the course of the disease, thus possibly implying that autoantibody should be monitored in RA, and must get a stronger consideration in therapeutic decisions.
References
Footnotes
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.
Data sharing statement No additional data are available.