Article Text

Abstract
Objective Although Behçet's disease (BD) is a chronic inflammatory disorder of uncertain aetiology, the existence of familial BD with autosomal-dominant traits suggests that a responsibility gene (or genes) exists. We investigated a Japanese family with a history of BD to search for pathogenic mutations underlying the biological mechanisms of BD.
Methods 6 patients over 4 generations who had suffered from frequent oral ulcers, genital ulcers and erythaema nodosum-like lesions in the skin were assessed. Whole-exome sequencing was performed on genomic DNA, and cytokine production was determined from stimulated mononuclear cells. Inflammatory cytokine secretion and Nod2-mediated NF-κB activation were analysed using the transfected cells.
Results By whole-exome sequencing, we identified a common heterozygous missense mutation in A20/TNFAIP3, a gene known to regulate NF-κB signalling, for which all affected family members carried a heterozygous C243Y mutation in the ovarian tumour domain. Mononuclear cells obtained from the proband and his mother produced large amounts of interleukin 1β, IL-6 and tumour necrosis factor α (TNF-a) on stimulation as compared with those from normal controls. Although inflammatory cytokine secretion was suppressed by wild-type transfected cells, it was suppressed to a much lesser extent by mutated C243Y A20/TNFAIP3-transfected cells. In addition, impaired suppression of Nod2-mediated NF-κB activation by C243Y A20/TNFAIP3 was observed.
Conclusions A C243Y mutation in A20/TNFAIP3 was likely responsible for increased production of human inflammatory cytokines by reduced suppression of NF-κB activation, and may have accounted for the autosomal-dominant Mendelian mode of BD transmission in this family.
- Behcet's disease
- Cytokines
- Inflammation
- Fever Syndromes
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
Behçet's disease is a chronic inflammatory disorder of uncertain aetiology. Familial Behçet's disease inherited in an autosomal-dominant manner does exist, but the pathogenesis remains unknown.
A20/TNFAIP3 gene mutation reinforces inflammation in humans and causes autosomal-dominant Behçet's disease.
Since A20/TNFAIP3 regulates NF-κB signalling, we can explain the curative effect of glucocorticoids, which are potent inhibitors of NF-κB activation.
Introduction
Behçet's disease (BD) is a chronic inflammatory disorder of unknown aetiology, characterised by recurrent oral aphthous ulcers, genital ulcers, uveitis and erythaema nodosum (EN)-like lesions on the skin.1 Involvement of the gastrointestinal tract and central nervous system as a subtype can be life-threatening.1 Earlier generational studies have proposed that some families inherit BD in an autosomal-dominant or recessive manner.2–4
Also referred to as tumour necrosis factor α-induced protein (TNFAIP) 3, A20 was first identified in endothelial cells as a primary response gene induced on tumour necrosis factor (TNF) stimulation.5 ,6 A20 was shown to be a ubiquitin-editing enzyme containing aminoterminal deubiquitinating activity mediated by its ovarian tumour (OTU) domain,7 which controlled NF-κB signalling by deubiquitinating receptor-interacting protein (RIP) 1, RIP2 and TNF receptor-associated factor (TRAF) 6.8 ,9 Multiple genetic studies have identified A20/TNFAIP3 as a susceptibility locus in inflammatory disorders,10 including rheumatoid arthritis,11 systemic lupus erythaematosus, inflammatory bowel disease (IBD) and BD,12 ,13 in addition to multiple B cell lymphoma.14 A significantly increased prevalence of TNFAIP3 polymorphisms has been reported in Chinese patients with BD.12 However, the polymorphisms are not caused by a non-synonymous mutation; they are located in the non-coding region, and do not have an increased prevalence in the European population.13 Furthermore, the NF-κB inhibitor A20 is a ubiquitin-modifying enzyme that might be critical in regulating human inflammatory diseases by inhibiting interleukin 1β synthesis,15 and reduced expression was associated with IBD and other kinds of spontaneous chronic inflammation in a murine system.16 Interestingly enough, myeloid-specific A20-deficiency in mice results in spontaneous development of a severe destructive rheumatoid arthritis,17 which crucially relies on the NLRP3 inflammasome-mediated caspase and IL-1β secretion.18 Most recently, Zhou et al19 reported six unrelated families with early-onset systemic inflammation resembling BD, which is caused by high-penetrate heterozygous mutation in A20/TNFAIP3.
In the present study, we employed whole-exome sequencing, analysis of inflammatory cytokine production in mononuclear cells and transfected cells, and luciferase reporter assays, to search for pathogenic mutations in a family with apparently autosomal-dominantly transmitted BD.
Patients and methods
Patients and family history
The proband (patient 1) was a 17-year-old Japanese boy who was referred to our hospital with a 5-month history of recurrent painful oral ulcers accompanied by fever, in November 2014. Oral ulcer with fever occurred once every 1–2 weeks, each time lasting for 1–2 weeks. The patient had suffered from frequent oral ulcers from the age of 9 years. He also had a 1-month history of unknown fever when 13 years of age and nephrotic syndrome 2 years later. Since the nephrotic syndrome had relapsed five times after discontinuation of steroid treatment, he was being treated with cyclosporine (175 mg/day) and mizoribine (250 mg/day) as maintenance therapy. Physical examination revealed painful oral ulcers, EN-like lesions on lower extremities, pseudofolliculitis on the trunk and a large painful ulcerative lesion in the perianal area. Laboratory examination revealed white cell counts 9470/μL, neutrophil rate 90.7%, haemoglobin 13.3 g/dL and C reactive protein 0.27 mg/dL (normal <0.1 mg/dL). Serum concentrations of IgG, IgA, IgM and IgD were 1623 mg/dL, 574 mg/dL, 104 mg/dL and 1.3 mg/dL, respectively. Although tests for autoantibodies and HLA-B51 were negative, a skin pathergy examination was positive. A clinical diagnosis of BD was made based on the International Study Group Criteria for BD (criteria for the diagnosis of BD).20 The oral ulcers were refractory to colchicine (1.5 mg/day), but responded promptly to the addition of low doses of prednisolone (15 mg/day).
Including this patient, six patients with BD have existed over four generations in the proband's family (figure 1). Patient 2, the 43-year-old mother of the proband, had also suffered from recurrent oral ulcers from the age of 8 years. She had been diagnosed as having BD due to recurrent oral and genital ulcers, EN-like lesions and pseudofolliculitis when she was 20 years of age. Colchicine treatment was unsuccessful. At 28 years of age, entero-BD was diagnosed based on gastrointestinal symptoms and endoscopic findings of intestinal ulcers, and responded to prednisolone (45 mg/day) and was thereafter controlled by low-dose steroids. Patient 3, the 71-year-old grandmother of the proband, had suffered from recurrent oral ulcers from the age of 8 years. Oral and genital ulcers, EN-like lesions and pseudofolliculitis with fever manifested frequently, but were resolved by low-dose glucocorticoids. Patient 4, a 42-year-old maternal aunt of the proband, had experienced oral ulcer from the age of 10 years. She had been diagnosed as having BD based on recurrent oral and genital ulcers, EN-like lesions and pseudofolliculitis, which responded poorly to a single dose of colchicine. Patient 5, an 18-year-old maternal female cousin of the proband, had experienced recurrent episodes of self-resolving oral and genital ulcers, EN-like lesions and pseudofolliculitis, since the age of 12 years. Patient 6, the proband's maternal great-grandmother, had died at 88 years of age. She had also suffered from recurrent oral and genital ulcers, and indurated erythaema accompanied by fever, from elementary school age, in spite of colchicine treatment. A history of genital ulcers could not be ascertained. None of the patients had exhibited ocular lesions of BD.

Family tree and A20/TNFAIP3 mutations. Sequence analysis of the A20/TNFAIP3 gene among the proband and family members revealed a heterozygous mutation of C243Y in exon 5, which was absent in the healthy younger brother. Sequence analysis was not possible on patient 6. Sequence analysis results are shown using reverse primer.
Apart from IL-1β and TNF-α (both <10 pg/mL), serum inflammatory cytokine levels were increased in the proband at presentation (IL-6: 134.1 pg/mL, IL-8: 83.1 pg/mL, IL-10: 12.1 pg/mL, granulocyte colony-stimulating factor (G-CSF): 184.8 pg/mL and interferon g: 22.2 pg/mL), but became persistently normal following the administration of glucocorticoids.
Whole-exome sequencing
Whole-exome sequencing was conducted on genomic DNA extracted from mononuclear cells from the proband (patient 1) and his mother (patient 2) (Takara Bio Inc, Mie, Japan). We prepared DNA libraries from 2.0 μg of genomic DNA, using a Paired-End DNA Sample Preparation Kit (Illumina, San Diego, California, USA). DNA was fragmented using Covaris technology, and libraries were prepared. We performed target enrichment, using a SureSelect Human All Exon V5 Kit (Agilent Technologies, Santa Clara, California, USA). Captured DNA libraries were amplified using supplied paired-end PCR primers. Sequencing was performed with an Illumina HiSeq 2500. We mapped the provided read sequences using BWA-MEN (0.7.10-r789). Alignment with the Genome Reference Consortium human reference 37 was performed with GeneData Expressionist software.
Mutation analysis
Heparinised blood from all affected members, apart from patient 6, as well as from the proband's healthy younger brother, was collected for genetic analysis, after obtaining informed consent. DNA was extracted from the samples, using standard methods. Direct sequencing of the A20/TNFAIP3 gene was performed using primers, as reported previously.21
Cytokine assay by mononuclear cells
Purified mononuclear cells were incubated in 96-well culture plates (0.5–1×105 cells/well) with medium alone, indicated concentrations of LPS (Sigma-Aldrich, St Louis, Missouri, USA), 25 μg/mL of poly (I:C) (InvivoGen, San Diego, California, USA), 1 μg/mL of CpG complementary DNA (cDNA), or 10 ng/mL of N-acetylmuramyl-L-alanyl-D-isoglutamine (muramyl dipeptide (MDP); Sigma-Aldrich) cultured for 18–48 h. The CpG oligodeoxynucleotide 2006 was purchased from Sigma-Aldrich. Supernatants were analysed for cytokine production, using cytometric bead array (CBA) Kits (BD Biosciences, San Diego, California, USA). Serum cytokine concentrations were additionally assayed by CBA Flex Set (BD Biosciences).
Measurement of cytokine secretion from THP-1 cells
Expression plasmids encoding C243Y or wild-type (WT) A20 were constructed, as reported previously.22 Monocytic leukaemia THP-1 cells were cultured in RPMI 1640 (Life Technologies, Carlsbad, California, USA), 10% heat inactivated fetal bovine serum (FBS), penicillin and streptomycin. An Amaxa Nucleofector (Amaxa, Cologne, Germany) was used to transfect 1×106 cells with 1 µg of pcDNA3, pcDNA3-A20, or pcDNA3.1-C243Y A20 together with 100 ng of pGL4.74[hRluc/TK] (Promega, Madison, Wisconsin, USA), as described by the manufacturer's protocol. In some experiments, 1 and 100 ng/mL LPS (Sigma-Aldrich) were employed to treat the THP-1 cells. Eight hours after medium replacement, the concentrations of IL-1β, IL-6, IL-8 and TNF-α in culture supernatants were measured by ELISA (BD Biosciences). Final concentrations were calculated by normalisation with the activity of the Renilla cotransfected reporter vector (pGL4.74[hRluc/TK]).
NF-κB assay
Human embryonic kidney (HEK) 293T cells were maintained in DMEM (Gibco) with 10% heat inactivated FBS, penicillin and streptomycin. Transfection was carried out with transfection reagent (Roche, Mannheim, Germany). 1×105 HEK293T cells were cotransfected with expression plasmids in the presence of 0.03 or 0.3 μg of reporter plasmids (pcDNA3, pcDNA3-A20 or pcDNA3.1-C243Y A20), 33 ng of pcDNA3-Nod2-Flag and 33 ng of pcDNA3-RICK-myc together with 8.3 ng NF-κB-dependent pBxVI-luc reporter and 8.3 ng of pGL4.74[hRluc/TK]. NF-κB luciferase reporter activity was measured 24 h post-transfection and values were normalised to those of firefly luciferase to Renilla luciferase activity.
Detection of NLRP3 transcripts
Total RNA was extracted from 2×106 mononuclear cells obtained from patient 1, normal individuals and THP-1 cells transfected expression plasmids encoding C243Y or WT A20 with or without 1 ng/ml of lipopolysaccharide (LPS) stimulation, with a TRIzol rapid RNA purification kit (Life Technologies, Grand Island, New York, USA). First-strand cDNA copies were synthesised using SuperScript II Reverse Transcriptase (Invitrogen, Carlsbad, California, USA) with oligo (dT) (Invitrogen, San Diego, California, USA) as the primer in a total volume of 20 μL. The following oligonucleotide primers were used: for β2-microglobulin (β2-MG), 5′-ACCCCCACTGAAAAAGA-3′ and 5′-CTCCTAGAGCTACCTGTGGAGCA-3′, and for NLRP3, 5′-GCCACGCTAATGATCGAC-3′ and 5′-TGCACAGGCTCAGAATGCTC-3′ sense and antisense, respectively. A quantity of 2 μL cDNA was amplified using each primer and Taq DNA polymerase by 35 cycles of the following steps: denaturation (94°C, 30 s), annealing (57°C, 30 s) and elongation (72°C, 60 s). The final polymerisation step was extended for another 5 min. The amplified products were analysed on a 1.2% agarose gel and visualised by ultraviolet light illumination. The relative integrated optical density of the messenger RNA (mRNA) bands was estimated with image processing and analysis in Java.
Results
Heterozygous C243Y mutation of A20/TNFAIP3 in patients with autosomal-dominant BD
Whole-exome sequencing was performed on a Japanese boy (patient 1) born to non-consanguineous parents, and on his mother (patient 2) (figure 1). The analysed variants were then filtered. Given the family history, we sought for defects with autosomal-dominant inheritance. The first filter, therefore, selected mutations present in a common heterozygous state between patients 1 and 2 in non-synonymous coding, identifying an initial pool of 1311 variants. The second filter selected a frequency of mutated nucleic acid between 0.4 and 0.6. Since we considered that the gene variants had an extremely high penetration rate, we assumed that the frequency of normal individuals carrying the pathogenic mutation was extremely low levels. Thus, we then focused on variants with <0.1% frequency in the Japanese allele frequency data (HGVD Release V.1.42: http://www.genome.med.kyoto-u.ac.jp/SnpDB/) and East Asia allele frequency data (ExAC release V.0.3: ftp://ftp.broadinstitute.org/pub/ExAC_release0.3/ExAC.r0.3.sites.vep.vcf.gz). This resulted in a final set of 43 variants of 32 different genes. TNFAIP3, CASP8-associated protein (CASP8AP2) and Wnt inhibitory factor 1 precursor (WIF1) were the only genes from this set known to be related to inflammation. We focused on the mutation of TNFAIP3, also known as A20, on chromosome 6, because A20/TNFAIP3 gene analysis data in multiple genetic studies, functional analyses and the results of knockout mice, have been extremely similar to those in BD pathogenesis.16 ,18 ,21–23 Sequence analysis of the coding and non-coding exons of A20/TNFAIP3 revealed the presence of a heterozygous C243Y mutation (728 G>A) in exon 5 in addition to three nucleotide deletions in an intron of upper exon 3 in the proband. The deletion was not found in the mother (patient 2), suggesting that they were paternally derived (not investigated). Meanwhile, the heterozygous C243Y mutation was present in all familial patients with BD examined, but absent in the healthy sibling and 64 unrelated normal controls of the same Japanese ethnicity. The identified C243Y mutation has been documented in neither the 1000 Genomes project data set (http://www.1000 genomes.org) nor in the Exome Variant Server data base (http://evs.gs.washington.edu/EVS/). Thus, the (chr6_138197226_G>A, C243Y) mutation of TNFAIP3 was suspected to be the gene responsible for autosomal-dominant BD.
Hyperproduction of inflammatory cytokines
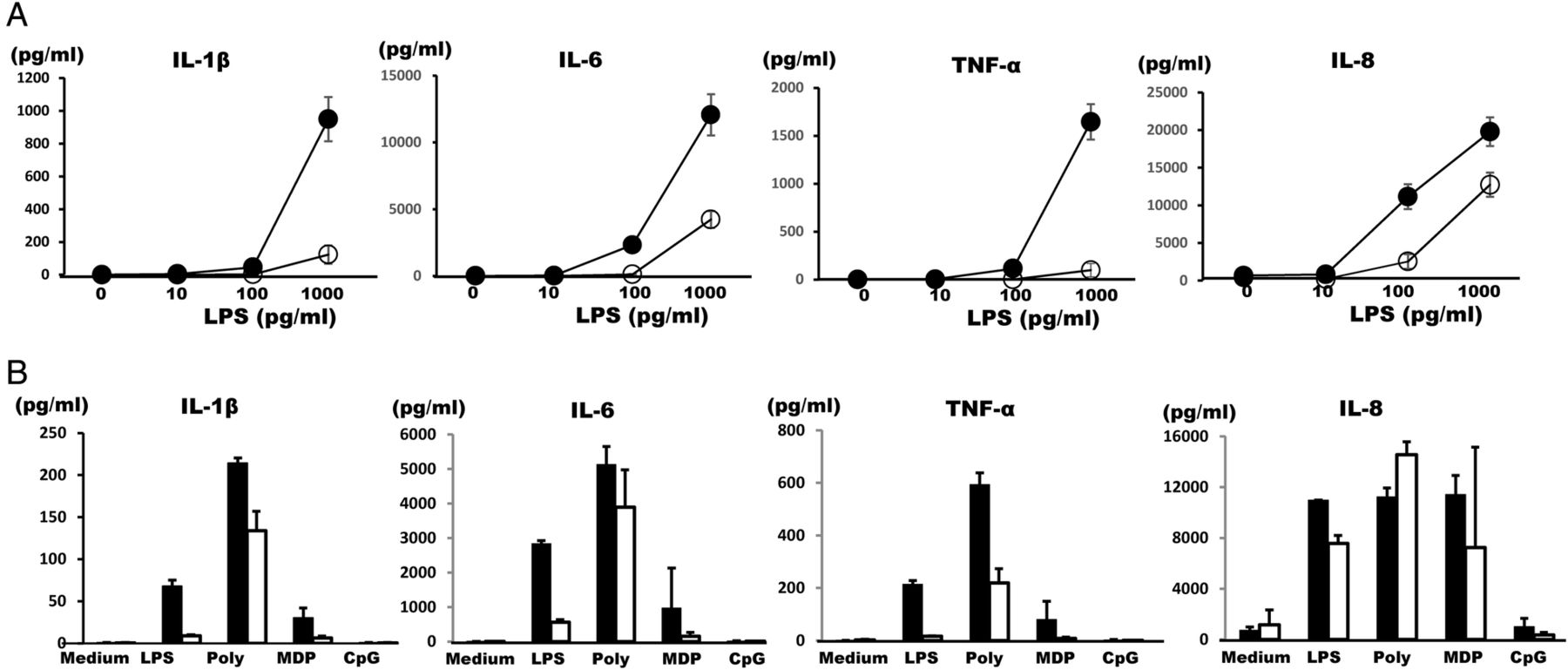
Duong et al15 reported that bone marrow-derived macrophages from tnfaip3−/− mice secreted IL-1β in response to toll-like receptor (TLR) ligands, LPS, or poly (I:C), but those from WT mice exposed to TLR ligands alone did not do so. Similarly to those from the tnfaip3−/− mice, the mononuclear cells obtained from patient 1 produced large amounts of IL-1β, IL-6 and TNF-α on LPS at various concentrations as compared with normal controls, although little difference was seen for IL-8 (figure 2A). Spontaneous production of these inflammatory cytokines was not observed with medium alone (figure 2A). Mononuclear cells obtained from patient 2 also produced remarkable amounts of IL-1β, IL-6 and TNF-α with LPS, MDP, or poly (I:C), but not with CpG DNA or TNF-α (data not shown), as compared with her son with neither BD symptoms nor A20/TNFAIP3 mutation (figure 2B). No marked differences were noted for IL-8 (Figure 2B). Thus, hyperproduction of inflammatory cytokines was observed in the patients.

Cytokine synthesis from mononuclear cells. (A) Mononuclear cells obtained from patient 1 (●) and healthy control (○) were cultured with the indicated concentrations of LPS for 24 h, after which supernatant cytokine concentrations were measured using CBA kits. The results of triplicate experiments are shown as mean±SD. One representative result of three independent experiments is shown. (B) Production of IL-1β, IL-6, TNF-α and IL-8 by mononuclear cells obtained from patient 2 (▪) and the healthy younger brother of the proband (□). Mononuclear cells were cultured with medium alone, LPS (100 pg/mL), poly (I:C) (25 μg/mL), MDP (10 ng/mL), or CpG DNA (1 μg/mL) for 24 h. Cytokine concentrations in culture supernatants were measured using CBA kits. The results of triplicate experiments were expressed as mean±SD. The data shown are representative of two independent experiments with different healthy controls, and one representative result is shown. CBA, cytometric bead array; IL-1β, interleukin 1β; LPS, lipopolysaccharide; MDP, muramyl dipeptide; TNF-α, tumour necrosis factor α.
Inflammatory cytokine secretion from THP-1 cells was suppressed by WT A20 but suppressed to a lesser extent by mutated C243Y A20
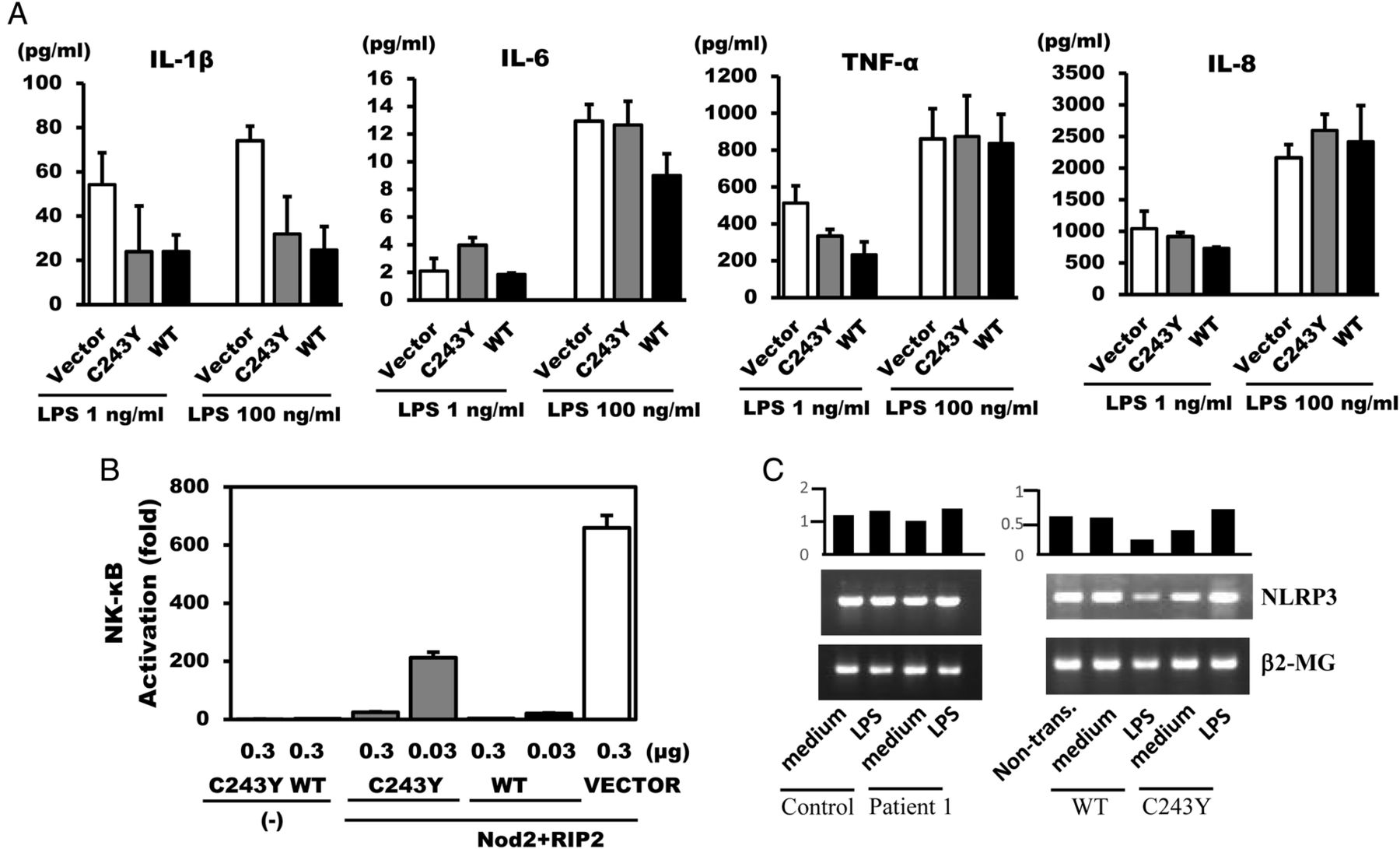
To confirm that the mutated A20/TNFAIP3 was responsible for the hyperproduction of inflammatory cytokines, we ectopically expressed the mutant and WT forms of A20/TNFAIP3 in THP-1 cells that had been transfected with pcDNA3.1, pcDNA3.1-WT A20, or pcDNA3.1-C243Y A20, together with pGL4.74 [hRluc/TK]. Transfection efficiency was normalised using Renilla luciferase activity. In THP-1 cells transfected with pcDNA3.1-WT A20, IL-1β secretion was significantly suppressed, IL-6 was suppressed in the presence of 100 ng/mL LPS and TNF-α was suppressed in the presence of 1 ng/mL LPS, but less so with regard to IL-8. Compared with the remarkable reduction in cytokine secretion from THP-1 cells by pcDNA 3.1-WT A20 transfection, IL-1β, IL-6 and TNF-α secretions were markedly much less suppressed by pcDNA 3.1-C243Y A20 transfected THP-1 cells at specific concentrations of LPS (figure 3A). These data indicated that the restraint of inflammatory cytokine secretion by A20 carrying the C243Y mutation was attenuated in comparison with that by WT A20.

Inflammatory cytokine secretion from THP-1 cells, luciferase reporter assay of Nod2-mediated NF-κB activation and NLRP3 expression. (A) THP-1 cells were transfected with vector control (□), pcDNA3.1-C243Y A20 ( ), or pcDNA3.1-WT A20 (▪) expression plasmids together with pGL4.74 [hRluc/TK]. Eight hours after LPS stimulation, the concentrations of IL-1β, IL-6, IL-8 and TNF-α in the culture supernatants were measured using ELISA. One representative result of two independent experiments is shown. (B) HEK293T cells were cotransfected with vector control (□), pcDNA3.1-C243Y A20 (▪), or pcDNA3.1-WT A20 (▪) expression plasmids together with pcDNA3-Nod2-Flag, pcDNA3-RICK-Myc, NF-κB-dependent pBxVI-luc reporter and pGL4.74[hRluc/TK]. NF-κB luciferase reporter activity was measured 24 h post-transfection. One representative result of two independent experiments is shown. Transfection efficiency of THP-1 cells (A) or HEK293T (B) were normalised using Renilla luciferase activity generated by cotransfection of pGL4.74[hRluc/TK]. (C) Total RNA was extracted from patient 1, normal individuals and THP-1 cells transfected by C243Y or WT A20 with or without LPS stimulation, after 8 h incubation. The mRNA expression of NLRP3 was determined using RT-PCR analysis with β2-MG as a control. The same results were obtained with 14 h incubation. The hold induction based on β2-MG (NLRP3/β2-MG) is shown on each upper band. β2-MG, β2-microglobulin; HEK293T, human embryonic kidney 293T; IL-1β, interleukin 1β; LPS, lipopolysaccharide; RIP2, receptor-interacting protein 2; TNF-α, tumour necrosis factor α; WT, wild-type.
), or pcDNA3.1-WT A20 (▪) expression plasmids together with pGL4.74 [hRluc/TK]. Eight hours after LPS stimulation, the concentrations of IL-1β, IL-6, IL-8 and TNF-α in the culture supernatants were measured using ELISA. One representative result of two independent experiments is shown. (B) HEK293T cells were cotransfected with vector control (□), pcDNA3.1-C243Y A20 (▪), or pcDNA3.1-WT A20 (▪) expression plasmids together with pcDNA3-Nod2-Flag, pcDNA3-RICK-Myc, NF-κB-dependent pBxVI-luc reporter and pGL4.74[hRluc/TK]. NF-κB luciferase reporter activity was measured 24 h post-transfection. One representative result of two independent experiments is shown. Transfection efficiency of THP-1 cells (A) or HEK293T (B) were normalised using Renilla luciferase activity generated by cotransfection of pGL4.74[hRluc/TK]. (C) Total RNA was extracted from patient 1, normal individuals and THP-1 cells transfected by C243Y or WT A20 with or without LPS stimulation, after 8 h incubation. The mRNA expression of NLRP3 was determined using RT-PCR analysis with β2-MG as a control. The same results were obtained with 14 h incubation. The hold induction based on β2-MG (NLRP3/β2-MG) is shown on each upper band. β2-MG, β2-microglobulin; HEK293T, human embryonic kidney 293T; IL-1β, interleukin 1β; LPS, lipopolysaccharide; RIP2, receptor-interacting protein 2; TNF-α, tumour necrosis factor α; WT, wild-type.
Diminished suppression of Nod2-mediated NF-κB activation by C243Y A20, and the effect on NLRP3 expression
A20 is a negative regulator of the inflammatory response,24 and downregulates Nod signalling by deubiquitination of RIP2.8 To assess if mutated A20 affects Nod2-mediated NF-κB activation through RIP2, WT or C243Y A20 was coexpressed in HEK293T cells with pBxVI-luc, pGL4.74 [hRluc/TK], pcDNA3-Nod2-Flag and pcDNA3-RIP2-myc, and NF-κB luciferase reporter activity was measured and the values were normalised to those of firefly luciferase to Renilla luciferase activity. Notably, A20 completely abolishes NF-κB signalling via Nod2 with only a small amount (33 ng), while the suppression of NF-κB activation by C243Y A20 was approximately two-thirds that of intact A20 and, therefore, a small amount. Interestingly, transfection with a large amount of C243Y A20 (333 ng) completely attenuated NF-κB signalling (figure 3B). This finding indicates that the A20 variant possessing the C243Y mutation should be regarded as a variant that does not cause complete loss of function, but, rather, causes weak function.
Recent studies in mouse macrophages lacking A20 suggest that A20 negatively regulates NLRP3 inflammasome signalling by suppressing production of NLRP3,18 and most recently demonstrated the same results in human.19 We therefore investigated NLRP3 transcripts in patient or THP-1 cells transfected with the C243Y A20 mutant. NLRP3 transcripts were recognised in the patient's mononuclear cells and THP-1 cells without stimulation. On stimulation, the patient's mononuclear cells did not display a remarkable increased expression of NLRP3 mRNA, but THP-1 cells transfected with the C243Y A20 displayed increase of the transcript (figure 3C).
Discussion
This study revealed that affected individuals—over four generations—of Japanese familial BD inherited in an autosomal-dominant manner carried a heterozygous C243Y mutation in the OTU domain of A20/TNFAIP3, which has been shown to regulate NF-κB signalling. Patient 1's mononuclear cells produced large amounts of inflammatory cytokines on stimulation, and the C243Y mutant of A20/TNFAIP3 attenuated the suppression of inflammatory cytokine syntheses by reduced suppression of NF-κB activation.
Patients 1 and 2 exhibited a very good response to glucocorticoid treatment in comparison with other cases of BD without dominantly inherited traits, although this may have been reflective of their poor response to colchicine. The elevated serum inflammatory cytokines in the proband at presentation became persistently normal soon after the administration of low-dose prednisolone. Since glucocorticoids are potent inhibitors of NF-κB activation via induction of the κB α inhibitory protein,25 it appears that gene alteration in this autosomal-dominant form of BD is associated with a NF-κB pathway. BD closely resembles Blau syndrome/early-onset sarcoidosis, which are Nod2 gene-associated chronic autoinflammatory diseases characterised by skin rash, arthritis and/or eye involvement, with non-caseating granulomata as their pathological hallmark.26 The granulomatous formation present in BD and Blau syndrome may be associated with increased Nod1-mediated/Nod2-mediated NF-κB signalling.23 ,27
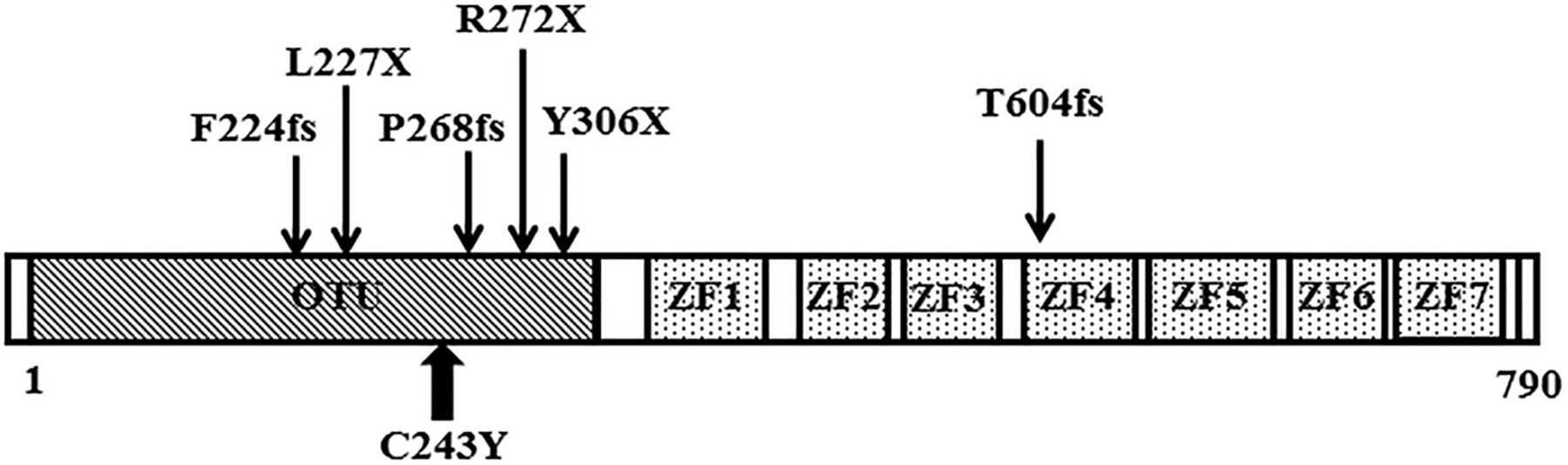
We focused on the A20/TNFAIP3 gene in this autosomal-dominantly transmitted BD family, on the basis of whole-exome sequencing data, A20/TNFAIP3 mutation analysis of the family members, functional analyses with transfectants and similarities with knockout mice data.12 ,15 ,16 A20 is an ubiquitin-editing enzyme containing aminoterminal deubiquitinating activity mediated by its OTU domain that removes Lys63-linked ubiquitin chains from RIP1/RIP2.7 As the C243Y mutation was within the OTU domain, A20-mediated inhibition by removal of Lys63-linked polyubiquitin chains from TRAF6 or RIP1/RIP2 may have been causative in this family's BD. These observations were comparable to those of knockout mice with an A20 deficiency that displayed highly secretion of IL-1β after stimulation with LPS or poly (I:C).15 Most recently, Zhou et al19 reported that heterozygous mutations of A20/TNFAIP3 were involved in early-onset autoinflammatory disease. Similar to our report, the disease resembled BD manifestations and all dominant mutations were located in OTU domains (one case possessed de novo mutation in zinc finger domain) (figure 4). The families with BD-like manifestations, described by Zhou et al, were of European or Turkish origin. Our family's is the first report of an A20/TNFAIP3 gene mutation in BD-like symptoms in patients of Asian ancestry. They also clearly demonstrated increased NF-κB activation, increased expression of proinflammatory cytokines and defective removal of Lys63-linked ubiquitin from TRAF6, NEMO and RIP1 in A20/TNFAIP3 mutated cells. We also found attenuation of the suppression of RIP2-mediated NF-κB signalling.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Domain structure of A20. The N-terminal ovarian tumour (OTU) domain is essential for deubiquitinase activity and C-terminal zinc finger (ZF1-7) domains mediate E3 ubiquitin-ligase activity. The location of the present mutation is represented with a bold up arrow and recently published mutations are indicated with down arrows.
We observed elevated levels of inflammatory cytokine synthesis in stimulated cells expressing A20 C243Y as compared with WT (figure 3A). In this experiment, it is important to assess the amount of exogenous A20 provided compared with that of endogenous A20, which presents normally in THP-1 cells. In our transfection system, transfection efficiency was normalised using Renilla luciferase activity. Therefore, we think that the exogenous amount of WT A20 and C243Y A20 is equal after transfection. We guess that the limited difference seen in figure 3A probably arises by the effect of additional WT A20. Since the polyubiquitination of RIP2 was essential for Nod1-mediated/Nod2-mediated NF-κB activation,8 we assessed the ability of mutant A20 C243Y proteins to suppress NF-κB, and found that the variant influenced Nod2-induced NF-κB activation. Considering the results of the luciferase reporter assay, it was interesting that A20 C243Y did not suppress Nod2-induced NF-κB activation completely, as did the WT, but, rather, acted in an apparently dose-dependent manner, indicating that this variant did not cause a complete loss of suppressive function. In contrast to Zhou et al's19 reported A20/TNFAIP3 mutations (nonsense or frameshift), C243Y might cause weaker loss-of-function. Myeloid-specific A20-deficient mice results show spontaneous development of severe destructive rheumatoid arthritis,17 which crucially relies on NLRP3 inflammasome-mediated caspase and IL-1β secretion.18 In our experiment, we could not confirm the enhanced NLRP3 mRNA, but could confirm elevated NLRP3 mRNA levels in C243Y A20 THP-1 cells on stimulation (figure 3C), in patient 1. The discrepancy in NLRP3 inflammasome activity in patients may depend on the individual variation. Given that NF-κB signalling cascades are strictly controlled by several proteins with ubiquitination, and that A20 modulates these processes, the slightly diminished impairment in suppressive function by C243Y may be responsible for persistent inflammation, and could lead to the development of BD.
In conclusion, genetic and functional analysis of familial BD disclosed that a heterozygous C243Y mutation in the OTU domain of A20/TNFAIP3 was responsible for the autosomal-dominant inheritance of BD in this Japanese family. The possibility to develop dominantly inherited BD by mutated A20/TNFAIP3, which controls NF-κB, supports the view that, at least, dominantly inherited BD can be regarded as an autoinflammatory syndrome.28 The accumulation of more family sets of BD cases and further molecular biological analysis of participation of polyubiquitination may shed light on the pathogenesis of autosomal-dominant BD and provide new clues on the mutation responsible for general BD.
Acknowledgments
The authors thank K Futagami, and D Tsuchiya for their help with gene analysis. They also thank Dr K Aya (Takara Bio Inc) for the support in whole-exome sequencing.
References
Footnotes
Contributors TS performed analyses of the whole-exome sequencing and of the patients. JM and KA incepted and designed the experiments, analysed the data and prepared the manuscript. Plasmid construction was prepared by Norimoto Kobayashi, cytokine assay by KK and analysis using mutated A20 was performed by Naoe Kaneko and NN. YT performed RT-PCR.
Funding This work was supported by a Health Labour Sciences Research Grant entitled ‘Translational research toward the clarification of autoinflammatory mechanisms by familial Mediterranean fever (FMF) inflammasomes based on the Mediterranean fever (MEFV) gene analysis (15ek0109033h0002)’.
Competing interests None declared.
Patient consent Obtained.
Ethics approval The MEFV and related gene analyses as well as cytokine assay were approved by the institutional review board of Shinshu University (authorisation number 447 and 476).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.