Article Text

Abstract
Giant cell arteritis (GCA) is one of the commonest forms of vasculitis in the elderly, and may result in blindness and stroke. The pathogenesis of GCA is not understood, although environmental, infectious and genetic risk factors are implicated. One gene of interest is PTPN22, encoding lymphoid protein tyrosine phosphatase (Lyp), expressed exclusively in immune cells, which is proposed to be an ‘archetypal non-HLA autoimmunity gene’. The minor allele of a functional PTPN22 single nucleotide polymorphism (rs2476601, R620W), which disrupts an interaction motif in the protein, was originally reported to be associated with biopsy-proven GCA in Spanish patients, with supporting data from three replicate Northern European studies. Recently, this observation was extended with additional patients and controls, and studies encompassing European, Scandinavian, UK and American patients. The aim of our study was to determine the association between PTPN22 rs2476601 (R620W) and biopsy-proven GCA in an Australian case cohort.
- Giant Cell Arteritis
- Gene Polymorphism
- Systemic vasculitis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
PTPN22 may be an archetypal autoimmunity gene.
In this study we have estimated the frequency of the PTPN22 rs2476601 (R620W) variant in Australian Giant Cell Arteritis (GCA) patients of European ancestry compared to population based controls.
Meta-analysis of all available studies from populations of both Northern and Southern European ancestry, confirm an association between the rs2476601 minor allele variant and GCA.
Giant cell arteritis (GCA) is one of the commonest forms of vasculitis in the elderly, and may result in blindness and stroke.1 The pathogenesis of GCA is not understood, although environmental, infectious and genetic risk factors are implicated. One gene of interest is PTPN22, encoding lymphoid protein tyrosine phosphatase (Lyp), expressed exclusively in immune cells, which is proposed to be an ‘archetypal non-HLA autoimmunity gene’.2 ,3 The minor allele of a functional PTPN22 single nucleotide polymorphism (rs2476601, R620W), which disrupts an interaction motif in the protein, was originally reported to be associated with biopsy-proven GCA in Spanish patients, with supporting data from three replicate Northern European studies.4 Recently, this observation was extended with additional patients and controls, and studies encompassing European, Scandinavian, UK and American patients.5 The aim of our study was to determine the association between PTPN22 rs2476601 (R620W) and biopsy-proven GCA in an Australian case cohort.
Australian patients with biopsy-proven GCA were recruited through the Arthritis Genomics Recruitment Initiative in Australasia (AGRIA), and the study consisted of 209 patients with biopsy-proven GCA and 455 South Australian European-ancestry population-based controls. The mean age of the patients with biopsy-proven GCA was 73 years, 66% were female, 70% presented with temporal headache, 1% with scalp tenderness, 54% with jaw claudication, 31% with visual disturbance, 4% with stroke and 40% with concurrent polymyalgia rheumatica.
rs2476601 Genotyping was performed using a Taqman allelic discrimination assay (C-16021387) on a Viia7 Real-Time PCR System (Applied Biosystems, Foster City, California, USA). A random-effects meta-analysis, based on the allele frequencies from all cohorts,5 was performed using the R library metafor.6
rs2476601 Genotypes and allele frequencies for Australian patients with GCA and controls are reported in table 1. The genotypes of the control population were in Hardy-Weinberg equilibrium (p=0.19). Although not reaching statistical significance, there was a trend towards an excess of minor (A) allele homozygosity within patients with GCA (p=0.09). There was a small increase in the minor (A) allele frequency in patients with GCA (OR 1.15, 95% CI 0.76 to 1.72), but this did not reach statistical significance (p=0.51)
Genotype distribution and minor (A) allele frequency of PTPN22 rs2476601 in Australian patients with giant cell arteritis (GCA) and controls
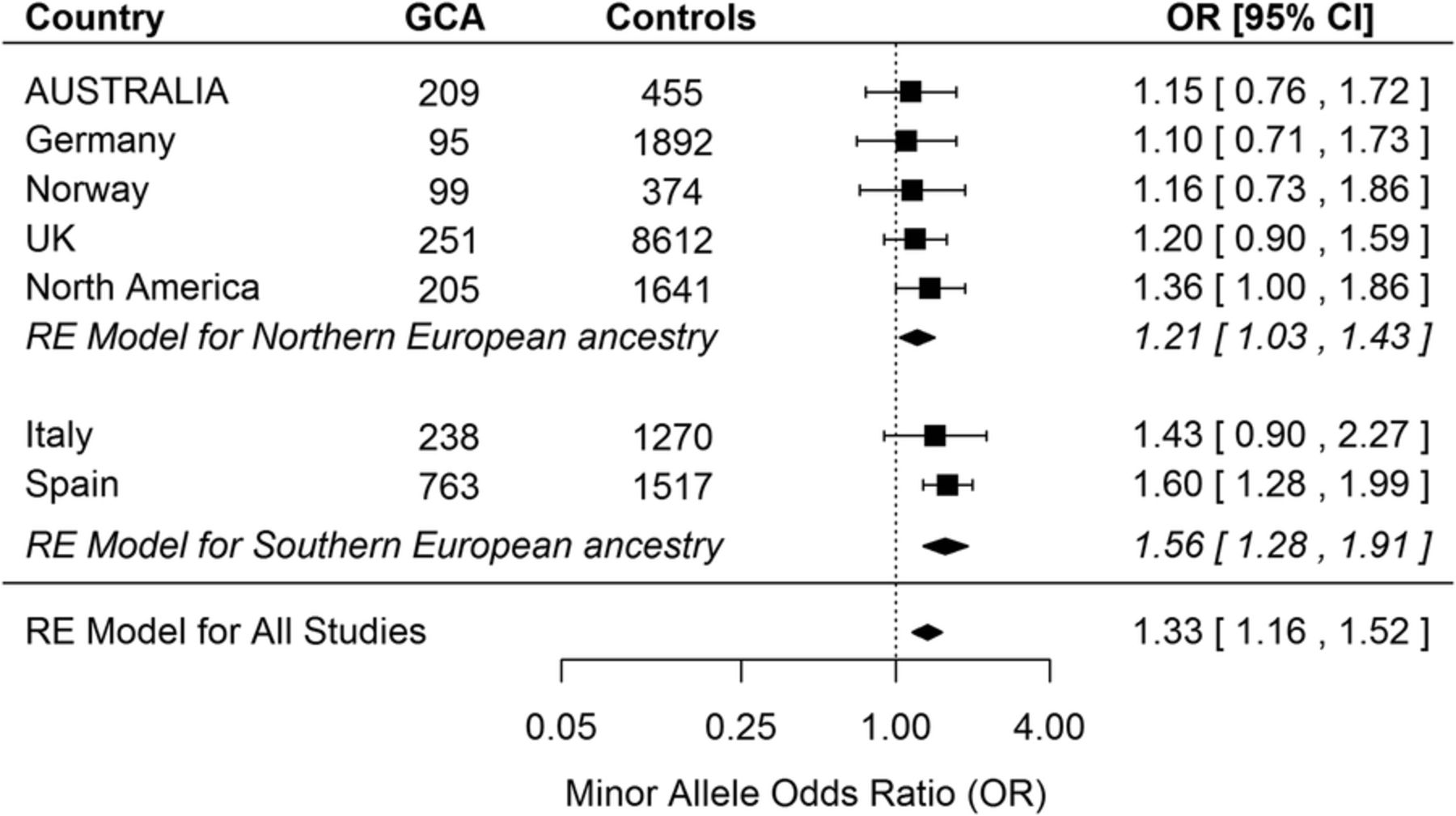
The meta-analysis of rs2476601 minor allele frequencies from all available GCA case–control studies is reported in figure 1. The effect size from our Australian study is in keeping with the findings from other studies of patients with Northern European ancestry. While the majority of these GCA studies, including ours, were underpowered to detect an association, the results from each study are remarkably consistent and collectively indicate a significant association between the rs2476601 minor allele and GCA (OR 1.33, 95% CI 1.16 to 1.52, p=3×10−5).

{kind=link}
Random-effects (RE) meta-analysis of PTPN22 rs2476601 minor (A) allele frequencies in patients with giant cell arteritis (GCA) (n=1860) compared to controls (n=15 761). Data from the current Australian study was combined with previously reported case–control studies.5 Results between the studies were consistent: τ2 was estimated as 0.0034 (on the log scale), and there was no substantive heterogeneity (Q=4.72, df=6, p=0.58). The difference in effect sizes between Northern and Southern European ancestry studies did not reach statistical significance (p=0.054).
Although not quite reaching statistical significance, there was also a trend towards larger effect sizes for studies involving Southern European ancestry participants (p=0.054). In this context, it is interesting that the risk of GCA, which is highest in Northern European populations (about 20 cases per 100 000 persons older than 50 years) and lower in Southern European populations (about 10 cases per 100 000),7 parallels a similar North-South cline in rs2476601 minor allele frequencies across Europe.3 Therefore, it is possible that PTPN22 rs2476601 minor allele contributes more to GCA risk on a non-Northern European genetic background, or alternatively, that GCA cases from Southern Europe have more Northern European ancestry than population-matched controls.
A limitation of this study is that the most appropriate genetic association model was not determined because full genotype information was only available for the Australian data. Interestingly, using the Australian data, the Akaike information criterion (AIC) suggested that the recessive association model (AIC 826.1) was slightly better than either the additive (AIC 830.7) or dominant models (AIC 831.1). However, given that only five minor allele homozygotes were observed in a total of 664 participants, there was insufficient information to determine the most appropriate model.
In summary, there is a significant association between the minor allele of PTPN22 rs2476601 and GCA, although the pathogenic mechanism remains to be elucidated. Power is a limiting factor in GCA genetic association studies due to difficulties in recruitment of elderly patients. However, this example illustrates the value of data from multiple, yet small, studies in delivering robust findings.
Acknowledgments
The authors wish to thank the Chief Investigators, study team, and participants of the North West Adelaide Health (NWAH) Study for the contribution of control samples for this study.
Footnotes
Contributors SL assisted with the conception and design of the work, genotyping, analysis and interpretation of the data, drafting the work and revising it critically for important intellectual content and gave final approval for the version published. AWH, LB, EDS, MDW, AH, GOL, TRM, TRM, BS, MDS, MR and MAB drafted the work and revised the manuscript. CDR conducted patient and control recruitment, analysed the data, drafted the work and revised the manuscript. GJ gave final approval for the version published. CLH assisted with patient and control recruitment, gave final approval for the version published, and substantial contributions to the conception and design of the work, drafting the work, revising the manuscript and final approval for the version published.
Funding National Health and Medical Research Council, Arthritis Australia (grant number 1068023).
Competing interests None declared.
Patient consent Obtained.
Ethics approval Human Research Ethics Committee (TQEH/LMH/MH).
Provenance and peer review Not commissioned, externally peer reviewed.
Data sharing statement No additional data are available.