Article Text

Abstract
Post-traumatic arthritis (PTA) develops after an acute direct trauma to the joints. PTA causes about 12% of all osteoarthritis cases, and a history of physical trauma may also be found in patients with chronic inflammatory arthritis. Symptoms include swelling, synovial effusion, pain and sometimes intra-articular bleeding. Usually, PTA recoveries spontaneously, but the persistence of symptoms after 6 months may be considered pathological and so-called chronic PTA. A variety of molecular, mechanobiological and cellular events involved in the pathogenesis and the progression of PTA have been identified. The activation of inflammatory mechanisms during the PTA acute phase appears to play a critical role in the chronic disease onset. Human studies and experimental models have revealed that a series of inflammatory mediators are released in synovial fluid immediately after the joint trauma. These molecules have been proposed as markers of disease and as a potential target for the development of specific and preventative interventions. Currently, chronic PTA cannot be prevented, although a large number of agents have been tested in preclinical studies. Given the relevance of inflammatory reaction, anticytokines therapy, in particular the inhibition of interleukin 1 (IL-1), seems to be the most promising strategy. At the present time, intra-articular injection of IL-1 receptor antagonist is the only anticytokine approach that has been used in a human study of PTA. Despite the fact that knowledge in this area has increased in the past years, the identification of more specific disease markers and new therapeutic opportunities are needed.
- Arthritis
- Inflammation
- Cytokines
- Osteoarthritis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Post-traumatic arthritis is a condition triggered by an acute joint trauma that can lead to osteoarthritis or chronic inflammatory arthropathies.
No feasible markers and specific treatments for preventing the evolution of post-traumatic arthritis in chronic disease are available yet.
What does this study add?
Inflammation occurring immediately after joint injury plays a key role in the onset of chronic post-traumatic arthritis.
How might this impact on clinical practice?
An early local anti-inflammatory therapy may represent an effective treatment option for preventing chronic post-traumatic arthritis.
Introduction
Joint injuries, with or without associated disruption of the articular surface, frequently lead to a progressive process of severe debilitating condition known as acute post-traumatic arthritis (PTA). PTA can occur at any age, in any joints and may develop from any kind of acute physical trauma, such as sports, vehicle accident, fall or military injury. Although a single trauma may sometimes be sufficient to induce arthropathy, repeated injuries and excess body weight are known to increase the risk for PTA.
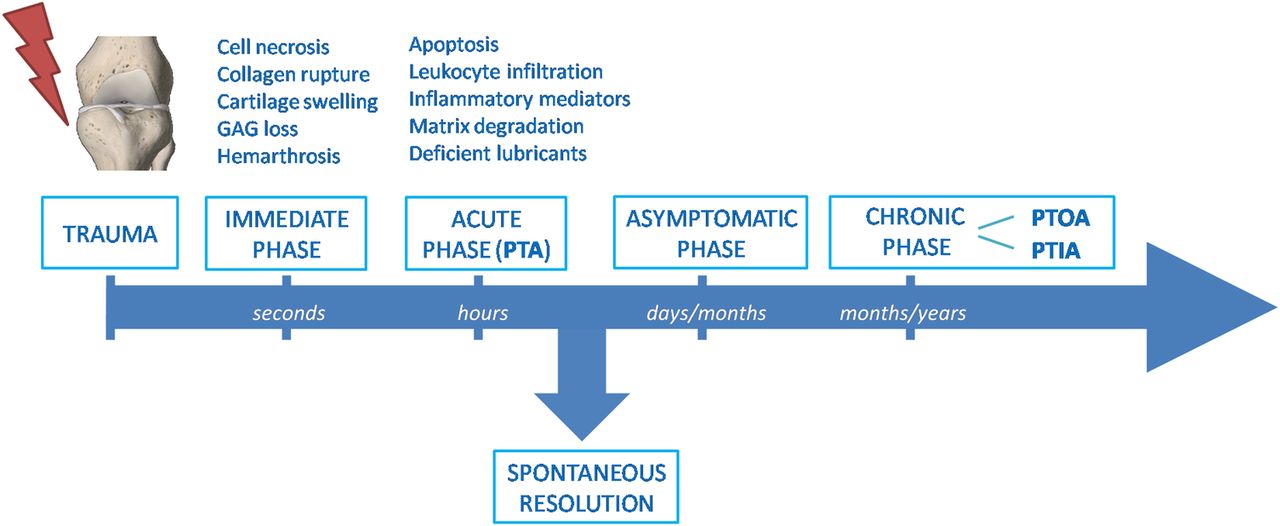
The acute symptoms following joint trauma include swelling, synovial effusion, severe pain and sometimes internal bleeding. Usually, PTA recoveries spontaneously in 2–3 months.1 The persistence of symptoms after this period should deserve attention and after 6 months, in clinical practice, it may be considered pathological and so-called chronic PTA. Chronic PTA can therefore represent an inflammatory condition that persists over time after a joint trauma. The most frequent chronic PTA is post-traumatic osteoarthritis (PTOA).1–4 However, a non-negligible number of PTA may lead to chronic inflammatory arthritis (PTIA), in particular psoriatic arthritis (PsA) (figure 1).5–8
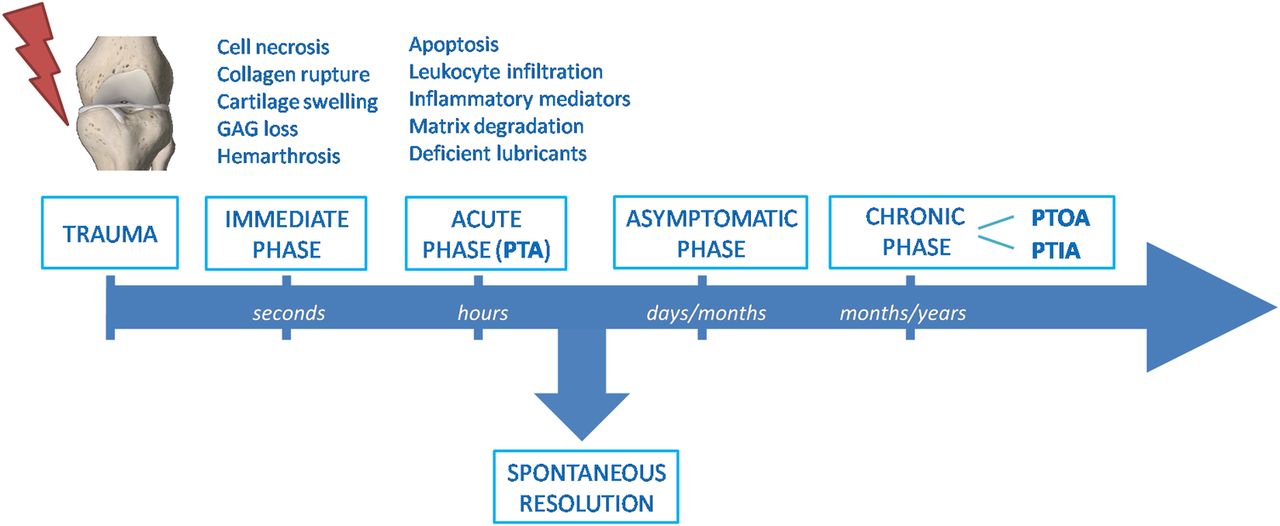
Timeline of the pathogenic processes following joint injury. After the immediate consequences of injury, mechanobiological, molecular and cellular changes in cartilage and other joint structures slowly progress into an acute post-traumatic phase. This inflammatory phase can spontaneously resolve after a couple of months or persist through a long clinically asymptomatic latency period. The chronic phase lasting years after the initial injury may lead to chronic OA or inflammatory arthritis. GAG, glycosaminoglycan; PTA, post-traumatic arthritis; PTOA, post-traumatic osteoarthritis; PTIA, post-traumatic inflammatory arthritis.
The pathogenesis of chronic PTA is not fully understood, and a variety of factors including genetic predisposition, epigenetic changes, mechanobiological and inflammatory mechanisms may be implicated.1 ,4 ,9 Despite the knowledge of the potential pathogenic mechanisms occurring in the injured joint has increased in the past years, specific predictive markers are lacking and current treatments of joint injuries fail to prevent evolution of PTA in chronic arthritis.
Post-traumatic osteoarthritis
Different studies show that joint injuries substantially increase the risk of OA, which further increases with patient's age at the time of injury and with time from the onset of injury.2 ,10 ,11 Other studies reported that ranges from 20% to more than 50% of patients who had had joint trauma develop OA and represent ∼12% of all OA cases.3 ,12
PTOA mainly affects younger adults than other forms of OA. Indeed, it has been reported that patients with disabling OA who had had an articular injury are more than 10 years younger than those who did not have joint trauma. In addition, 13.9% of patients with a history of joint injury during adolescence or young adults developed knee OA, compared with just 6% of those without a history of joint trauma.2
Common causes leading to PTOA include intra-articular fractures and meniscal, ligamentous and chondral injuries.13 The ankle is the most affected joint, accounting for 54–78% of over 300 000 injuries occurring in the USA alone each year.14 ,15 Knee injuries remain the most prevalent worldwide, with 700 000 cases annually in the USA and 12.5% of PTOA.16 ,17 However, even if less frequent, PTOA can occur also in other anatomical locations such as the shoulder and hip.18
Generally, PTOA is not clinically diagnosed until the onset of the symptomatic phase, which is highly variable. PTOA may occur early, in less than a year or remain asymptomatic for a long period of time, even 10–20 years after the trauma. However, in both cases, it is increasingly believed that the OA development in the injured joints initiates during the initial traumatic event by intra-articular pathogenic processes, such as apoptosis of articular chondrocytes, subchondral bone remodelling, cellular infiltration and the release of inflammatory mediators in synovial fluid (SF).
Post-traumatic inflammatory arthritis
A history of physical trauma may also be found in patients with chronic inflammatory arthritis with the percentage varying from 2% to 25%.5 Trauma is one of the most frequent causes of recurrence of acute attack in gout and pseudogout as a consequence of crystal shedding.19 ,20 Furthermore, it has been reported that patients who experienced significant joint injuries have an increased risk of developing calcium pyrophosphate and basic calcium phosphate crystal deposits.21 ,22 Different studies support the idea that a previous physical trauma may also be considered pathogenically relevant in other categories of inflammatory chronic arthritis, including rheumatoid arthritis (RA), juvenile chronic arthritis, juvenile or adult onset Still's disease and spondiloarthritis, especially PsA.23–26 It has been reported that the prevalence of trauma preceding arthritis was higher in patients with PsA than in patients with RA or ankylosing spondylitis.27 A preceding event was not found to be more common in patients with PsA who had positive human leucocyte antigen-B27 status. No difference was observed between post-traumatic and non-post-traumatic arthritis with regard to their clinical evolution.26
Several hypotheses have been suggested to explain the mechanisms behind the onset of PsA following joint trauma, but the exact pathogenesis is still not clear. A genetic predisposition, a ‘deep Koebner’ phenomenon and the release of self-antigens or neuropeptides, such as substance P and nerve growth factor, have been proposed.25 Alternatively, the concept of a synovium-entheses complex and an abnormal innate immunity pathway with an increased release of inflammatory cytokines has been supposed to play a key role in early post-traumatic PsA (PT-PsA). In this setting, entheses damage after injury may be identified as the initiator of the disease process and leads to the release of different proinflammatory substances, including interleukins, whose access to the adjacent synovium could trigger and perpetuate inflammation.8 ,28 Although all these potential local mechanisms following joint injury have been hypothesised to trigger the development of chronic inflammatory arthritis, it is difficult to define a link between the articular damage and causes of systemic events. No specific studies have been conducted to describe systemic consequences of joint trauma, even though an increase in the erythrocyte sedimentation rate (ESR) and in the serum C reactive protein (CRP) levels has been observed in post-traumatic spondyloarthropathy and PT-PsA.29–31
Pathogenic mechanisms and role of inflammation in early phase
The pathogenic mechanisms of post-traumatic arthropathies have been the subject of considerable researches in the past decade, improving the knowledge of the mechanobiological, molecular and cellular consequences of joint trauma. These processes can be temporally separated into three phases: the immediate phase related to mechanic events; the acute phase characterised by cell death/apoptosis and inflammatory events; and the chronic phase with joint pain and dysfunction (figure 1). The inflammatory acute phase can spontaneously resolve after a couple of months or slowly progress through a long clinically asymptomatic latency period.1
Biomechanical changes in cartilage and other joint structures (meniscus, ligament, subchondral bone, synovial membrane) occur immediately after the injury. The initial pathological changes may vary depending on the severity of the tissue damage: low-intensity injuries commonly cause articular surface damages without bone fractures, which instead are displaced by high-intensity injuries.32 The immediate cartilage swelling is caused by collagen rupture and rapid glycosaminoglycans (GAG) loss. Collagen damage is an irreversible step in the progression to PTOA, since the rapid loss of tensile behaviour of the tissue could not be restrained by the fibrillated collagen network.33 Moreover, the continuous loss of GAG can predispose cells to necrosis. Resident chondrocytes are known to mediate cartilage homoeostasis and it seems evident that reduction in cellularity diminishes the capacity for repairing or regenerating the cartilage after trauma. The acute postinjury mechanical overload can rapidly cause intra-articular bleeding by rupture of blood vessels in the joint.34 Haemarthrosis, mostly following anterior cruciate ligament (ACL) rupture and meniscal tear,35 is an important factor in the pathogenesis of PTOA, due to a close relationship between bleeding episodes and cartilage damages. It also leads to synovial hypertrophy and triggers synovitis at later stages.
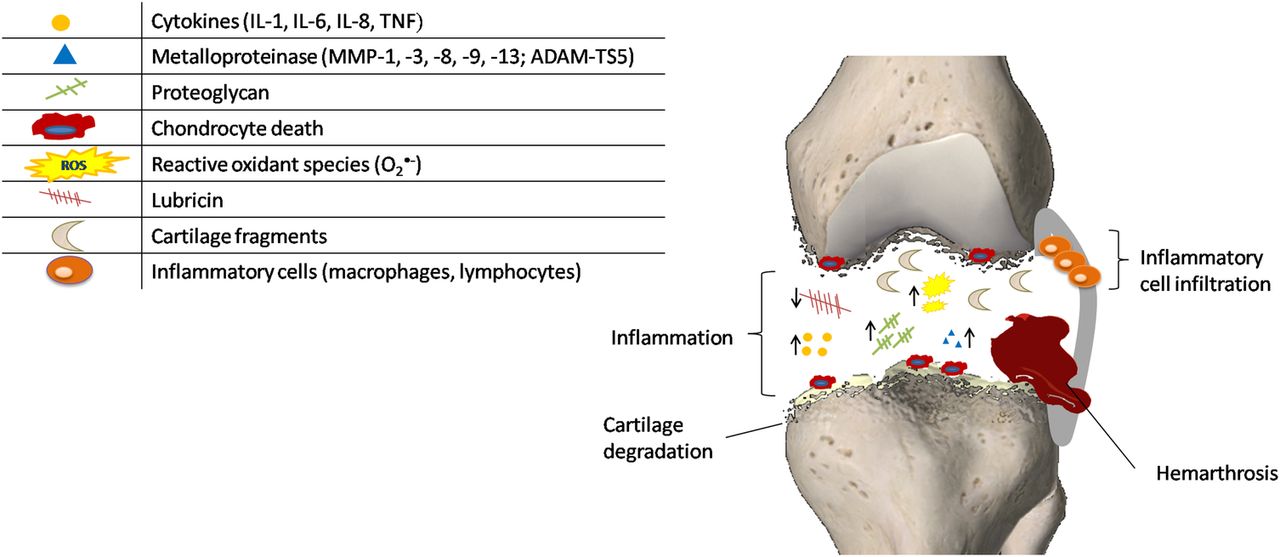
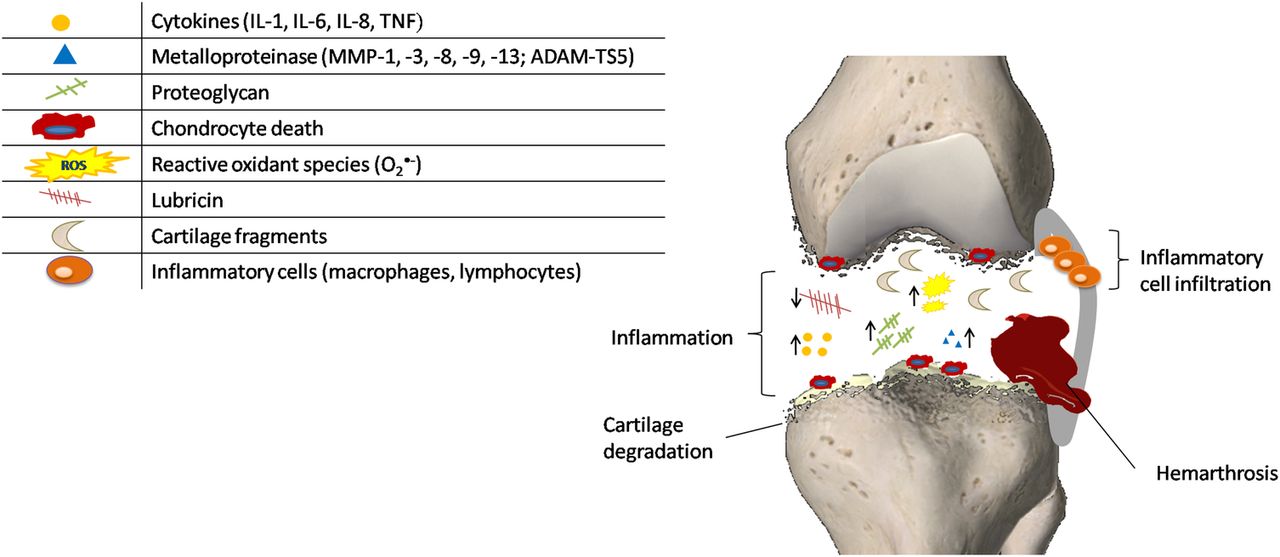
Complex metabolic changes respond to the prominent inflammation occurring among the acute post-traumatic phase lasting hours up to ∼2 months, as summarised in figure 2. Anderson et al36 observed three overlapping phases over the course of the first week or two after trauma: an early phase characterised by cell death and inflammatory events; a subacute phase with persistence of inflammation; and a late phase with increase in matrix degradation. The activation of complement proteolytic cascade and toll-like receptors (TLRs), such as TLR-2 and TLR-4, has been hypothesised to act in conjunction with the cytokine/chemokine network as the first line of defence of innate immunity.1 Evidence in human and animal articular cartilage suggested the activation of the remaining viable chondrocytes through the enhanced cell metabolism and the generation of oxygen radicals, matrix-degrading enzymes and inflammatory mediators.4 The exacerbation of the tissue biomechanical and physicochemical properties results in significant changes in chondrocytes, altering their ability to express proteins involved in metabolic pathways and leading to cell death. Since chondrocytes are responsible for maintenance of articular cartilage function, their death through apoptotic mechanisms is central in the development of PTOA. This is also supported by the fact that a higher percentage of apoptotic cells was found in the cartilage of patients with intra-articular fracture, compared to specimens of participants with OA and RA.37 ,38 Apoptosis may occur in the presence or absence of visible damage in cartilage, and it can be modulated by the caspase pathway.39 In vitro and in vivo study have identified a relationship between cell death and the following factors: impact energy, proximity to the articular surface and the presence of fracture.40–42 In particular, the presence of fracture has been shown to adversely affect chondrocyte viability and matrix metalloproteinase (MMP) and aggrecanase activity compared to joint impact without fracture.42 Controversial results have instead been reported with regard to the correlation between cell viability and time of the injury. In a study on human cartilage, chondrocyte apoptosis was found to be maximal at 5 days after intraarticular fracture.38 In contrast, a maximal state of apoptosis was described at 1–2 days after injury in human osteochondral fragments.43
{kind=link}
{kind=link}
Pathogenic mechanisms occurring during the immediate and acute phases after joint injury. The initial injury causes structural damage to the matrix of articular cartilage that evolves into several cellular responses ranging from the upregulation of matrix degradative pathways (suppression of collagen and proteoglycan synthesis, and overexpression of matrix-degrading enzymes), release of oxidants and inflammatory cytokines, and cell death. ↑=enhanced concentration of the component; ↓=decreased concentration of the component.
In addition to the continued GAG loss, injury to the cartilage allows the release or degradation of many other proteins as MMPs and collagen-type II.44 ,45 Many of these extracellular proteins come from the pericellular matrix and may be the result of higher turnover or damages in this matrix. Consistent with these findings, SF after injury presents a variety of matrix proteins and also shows elevated levels of fragments of collagen and cartilage oligomeric proteins generated by different aggrecanases. Since these fragments remain present for several years after the injury, they may contribute to the frequent development of OA following joint trauma.46 The lower concentration of the lubricants (hyaluronic acid and lubricin) observed in SF results in a severely compromised lubricating function. Besides the proteolysis by neutrophil-derived enzymes, the concentrations of these joint lubricants are also affected by inflammatory mediators.
Acute synovial inflammation associated with joint injuries is strictly related to cellular infiltration and correlated with the severity/extent of injury. Studies on animal models support a role for both infiltrating macrophages and T-lymphocytes in post-traumatic disease progression.47 As demonstrated in bovine explants, synovial inflammation also leads to oxidative damage to articular cartilage chondrocytes and the matrix via the increased secretion of reactive oxygen species (ROS) and reduced antioxidant defences.48 In addition to directly damaging chondrocyte viability, ROS synergise with proinflammatory cytokines and nitric oxide to promote catabolic gene expression via extracellular signal-regulated kinase (ERK)1/2 and c-Jun N-terminal kinase (JNK).
In certain individuals, the influence of anti-inflammatory factors (ie, IL-10 and IL-1 receptor antagonist) produced in the early postinjury period may allow resolution of inflammation and reduce the risk of progressing to PTOA. However, perpetuation of inflammation through a continued activation of innate inflammatory pathways, such as complement and danger signal-mediated pathways, may promote progression to chronic arthropathies.1 Moreover, the metabolic changes in articular cartilage gradually progress through a long clinically asymptomatic period to a structural deterioration and joint pain caused by chronic loading that can last years after the initial injury. In particular, subchondral bone remodelling, osteophyte development and fibrosis of the synovium or joint capsule may expand in this chronic phase the joint instability, increasing the risk of post-traumatic arthropathies. Studies of relationships between post-traumatic residual joint abnormalities and OA suggest that instability and articular surface incongruity following some severe acute joint injuries may lead to changes in chondrocyte metabolism and cartilage degradation.49 There is emerging evidence that inflammation and joint tissue remodelling lead to chronic responses of cartilage, as well as also fibrosis and failure of joint repair.50
Inflammatory mediators after joint injury
As discussed in the pathogenic mechanisms, it is not completely clear whether the primary cartilage damage results directly from mechanical trauma or from a process in the cartilage driven by biochemical substances.
Restoring knee-joint stability by ACL reconstruction has not been demonstrated to reduce the incidence of PTOA in most ACL injured knees, indicating that mechanical factors are not the unique determinants of OA.51
Other factors may therefore be responsible for early development of cartilage destruction and their detection in SF may have an extremely important diagnostic role but, even more, may represent prognostic biomarkers of OA and also of other joint diseases.
In a previous study, we demonstrated that about 10% of patients with PsA have a history of trauma to different joints (knee, shoulder, elbow, wrist, ankle or finger) before the onset of the disease. In particular, we observed that blood ESR, serum CRP and SF IL-6 levels were significantly higher in post-traumatic than in non-post-traumatic patients with PsA at disease onset but not after follow-up.26
Other studies on patients with knee injuries showed that SF contains increased levels of many inflammatory mediators immediately after the initial joint trauma. Numerous inflammatory cytokines are found at increased levels during the acute postinjury phase, including IL-1, IL-6, IL-8 and tumour necrosis factor (TNF). Lee et al52 reported that resistin, an inflammatory mediator derived from macrophages, may stimulate inflammatory cytokines release after injury.
In a population of patients with ACL tear, high levels of IL-1ß, IL-6 and IL-8 have been found in the SF collected within 48 hours after injury with respect to those collected later. Interestingly, they observed persistent lower levels of the IL-1 receptor antagonist (IL-1Ra) hypothesising an imbalance of this protecting factor induced by trauma at least at disease onset.53 The critical role of IL-1 in the acute phase of joint injury and, in particular, its local inhibition using IL-1Ra has also been clearly pointed out by Furman et al.54 They in fact demonstrated that a single intra-articular injection of IL-1Ra significantly reduced cartilage changes and synovitis after a joint fracture in mice.
Very recently, SF IL-6 upregulation in a population with acute knee injury has been strongly associated with clinical symptoms over the early period after injury.55
Raised SF concentrations of IL-6 after joint injury were also observed in other studies, along with those of monocyte chemoattractant protein (MCP)-1, macrophage inflammatory protein-1ß and interferon (IFN)-γ.56 ,57 These data are consistent with cytokine and proteinase expression patterns observed in animal models of joint injury, as discussed below, and indicate a postinjury activation of inflammatory pathways in cells of the synovium and cartilage.
Early signs of articular cartilage degradation, such as elevated MMPs production, collagen-type II peptide release, proteoglycan degradation and bone marker release, appear in the SF from patients following knee injury.58 MMP-3 and metalloproteinase with thrombospondin motifs (ADAMTS)-5 increased dramatically within 4 hours. The expression of MMP-13 is induced by collagen-II molecules and then act on the discoidin domain receptor 2 through mitogen-activated protein kinase signalling pathways (ERK and p38).59 Klatt et al60 also provide evidence that collagen-type II induces MMP-1, 2, 13, 14 as well as inflammatory cytokines IL-1β, IL-6 and IL-8.
It is known that joint injury compromises boundary lubrication of the SF. Lubricin, a chondroprotective mucinous glycoprotein, is considered to be the factor responsible for boundary lubrication of diarthrodial joints. Patients with a diagnosis of traumatic synovitis of the knee have been seen to exhibit a decrease in SF lubricating properties, reflecting decreased SF lubricin concentrations.61 Reduced lubricin levels have also been described in patients with ACL injury.62 In these participants, a relationship between the decrease in lubricin and an increase in proinflammatory cytokines IL-1ß, TNF and IL-6 has been observed in the early days post injury.63
Tenascin-C has been recently postulated to be a new marker of a local upregulation of inflammation pathways after joint injury.64 Tenascin-C is an extracellular matrix glycoprotein which, interacting with other matrix molecules, plays a major role in cell adhesion, migration, proliferation and cellular signalling. It is highly expressed in the cartilage and synovium of diseased or injured joints.65
Considering the low content of tenascin-C in normal adult articular cartilage, its marked increase in SF after injury has been postulated to be the product of an upregulated tenascin expression by chondrocytes and synoviocytes. It has therefore been considered a marker of a local upregulation of inflammation pathways. In particular, as an endogenous activator of the innate immune receptor TLR4, tenascin-C fulfils the criteria for damage-associated molecular patterns.66
In vitro and in vivo models
Many different mechanical and biochemical processes are involved in the initiation of PTA. It is therefore difficult to reproduce in vitro the tissue damage and to activate specific cellular pathways. Most of the studies investigate the role of trauma using tissue models of human cartilage and examining cell survival, gene expression and release of inflammatory mediators. Cartilage explants are submitted to a single impact loading or repetitive overload injury by means of different devices and the additive effect of cytokines,67 ,68 inhibitors or drugs on trauma-induced inflammatory process can be evaluated. Alternatively, engineered cartilage tissue analogues are produced using a suspension culture with biophysical properties similar to native articular cartilage and subjected to different injurious compression.69
Tissue damage, in particular cell death and matrix damage, depends strictly on the level of compression and the loading time.70
Animal models are crucial to understand the development of PTA and to assess novel possible therapies. Although mice are the most used, rats, guinea pigs, rabbits, cats, dogs, sheep, goats, mini pigs and pigs are also suitable.71 ,72 Experimental PTA is generally induced either through a surgical intervention, or by causing a physical trauma directly in the joint. In the first case, the knee patellar ligament is transected and the medial lateral menisci removed with a microsurgical technique. During the procedure, close attention has to be paid to not injure the articular cartilage. Alternatively, an osteotomy can be performed to disrupt the articular surface or the subchondral bone.73
The translational utility of the animal models used depends strictly on the selection of the most appropriate procedure to induce the joint damage and on the clinical outcome which has to be achieved.
For different reasons, such as the availability, the small size, the low cost and for not less important ethical issues, the mouse represents the most described animal model of PTA. The techniques used to induce PTA and the genetic background of the strains strictly influence the experimental outcomes. The surgical destabilisation of the medial meniscus (DMM) is currently the most widely performed procedure.74 DMM causes from mild to severe degenerative injures in the articular cartilage of the medial tibia within 10–12 weeks from the treatment, often associated with sclerosis of subchondral bone and mild synovitis.75 In the DMM mouse model, signs of inflammation occur very early and within 7–10 days from surgical procedure joint tissues and synovial fluid exhibit huge infiltrates of inflammatory monocytes and activated macrophages.76
Another helpful model of PTA is the transection or the disruption of the ACL. After ACL tear, the damages occurring in the joint are very similar to those occurring in clinical PTA: severe cartilage damage in the posterior tibia, significant subchondral erosion and synovium hyperplasia, and leucocyte infiltration within 8 weeks.71
ACL and DMM models show the highest levels of IL-1β, IL-6, IL-8 and MCP-1 at day 3 after injury. In the DMM model, MCP-1 appears especially upregulated at the transcriptional level within the first week, following a steady downregulation after 16 weeks from the procedure.77
Various studies demonstrate that, in the DMM and ACL mouse models, upregulation of inflammatory genes occurs very early after surgery but seems significantly downregulated after 3 months from injury according to data observed in human patients.1
Recently, the intra-articular fracture mouse model has been described by Furman et al. In this model, an intra-articular fracture is generated using an indenter tip driven by a software to apply a load to the anterior articular cartilage of the tibia.78 This intriguing model permits to observe the development of PTA after a fracture. Intra-articular fracture rapidly provokes severe erosion of the cartilage at the fracture site, significant bone loss and acute synovitis within 7 days.79
Despite the significant progress in the development of mouse PTA models, a consensus about the best procedure to use in translational studies is still lacking.72 Nonetheless, the animal models remain, at least for the moment, irreplaceable to reproduce the biological processes involved in PTA onset and for the development of new therapies.
Predictive markers for chronic PTA
Despite the knowledge of the potential, pathogenic mechanisms occurring in the injured joint have increased in the past years, whereas clinical and biochemical features that predict or correlate with the risk and progression of chronic PTA have to be still identified.
Given the high number of molecules involved in the processes during the acute injury phase, molecular biomarkers could be more useful to indicate the risk for chronic evolution. Indeed, molecular biomarker profiles based on the identities, quantities and temporal pattern of expression in joint tissues and, in particular, in SF may provide important indications of the disease development. At present, an increasing number of reports suggest that potential candidates could be inflammatory mediators, even though their value for clinical purposes has not yet been confirmed.
Recently, studies in animals have indicated that specific genetic mutations that alter the synthesis of a variety of molecules may also be relevant as predictive biomarkers for the pathophysiology of chronic PTA. In particular, modifications in genes involved in cartilage matrix degradation, inflammation,or the differentiation and apoptosis of chondrocytes contribute to protect against the onset of PTOA.80
Human studies in epigenetic phenomena may provide even more important information, but consistent results have not been achieved up to the present time. Disease progression may be facilitated by a decreased CpG methylation within the PH domain leucine-rich repeat-containing protein phosphatase 1 (PHLPP1) promoter that leads to an augmented PHLPP1 expression. PHLPP1 is a Ser/Thr phosphatase that decreases the activity of several kinases promoting cartilage anabolic signalling. Furthermore, PHLPP1 deficiency in mice surgically destabilised by transection of the medial meniscal ligament has been shown to protect against OA onset by increasing the cellular content and thickness of articular cartilage.81
Treatment and prevention
At present, there are no approved therapies to address the acute PTA and prevent the onset of the chronic disease. The primary goals of treating patients with PTA are to minimise the symptoms and loss of function and reduce pain. Currently, treatment for PTA includes anti-inflammatory drugs (non-steroidal anti-inflammatory drugs or intra-articular injections of cortisone), low impact exercise and lifestyle changes, for example, losing weight if necessary. However, not all patients benefit from the agent usually used and chronic arthritis can develop in the damaged region. Once the chronic disease has developed, the therapy is the same for the idiopathic forms. If none of these measures are effective, then surgery is the next option. However, any medical or surgical treatment can have severe side effects or risks.
An increased understanding of the molecular, mechanobiological and cellular events involved in the pathogenesis of chronic PTA may open interesting perspectives concerning new therapeutic opportunities and thereby offer patients safer and more effective drugs. Preventive measures are thought to be the most effective strategy to limit the degree of acute joint damage and the eventual development of chronic PTA. Thus, the ideal therapy should include early clinical interventions during the first phases after joint injury and address several pathogenic pathways.
A large number of molecules have been explored as potential targets for treatment in preclinical studies. Among these MMPs or caspase inhibitors, growth factors, antioxidants and even mesenchymal stem cells have shown an interesting effect as potential disease modifying drugs in PTA animal models.50 ,82
Since activation of inflammatory mechanisms is considered to be critical to development of chronic disease, anti-inflammatory interventions may represent the best available opportunity to intervene early in the acute post-traumatic period. A study carried out by Lewis et al supports this hypothesis using an animal model of the tibial plateau fracture. They observed that MRL/MpJ mice, which are known to have enhanced regenerative abilities in response to injury, exhibited lower levels of inflammation than wild-type mice, were protected from the progression of PTA.83 In particular, anticytokine therapy has demonstrated a marked efficacy as preventative agents of the long-term onset of chronic PTA. IL-1 inhibition, through knockout of IL-1β, intra-articular injection or adenoviral transfer of IL-1Ra and retroviral transduction to overexpress IL-1Ra, is resulted therapeutically effective in animal models of surgically induced PTA.74 ,84 ,85 Blocking of TNF increased the production of lubricin and decreased the release of GAG, resulting in a chondroprotective effect in a rat model of PTA.86 Recently, the use of lentiviral-mediated RNA interference silencing of IL-1b and TNF to treat PTA in rabbits displayed reduced cartilage damage and speed of degeneration.87 However, although both cytokines play a role in the post-traumatic acute phase, different studies performed in mouse models assert that intra-articular inhibition of IL-1, rather than TNF, may reduce the development of chronic PTA.54 ,88 ,89
Despite the use of all these agents has proven effective in reducing the progression of chronic PTA in animal models, only one small randomised pilot clinical trial has been conducted. Currently, IL-1Ra is the only agent that has been used as an anticytokine approach in patients with acute PTA. In this study, it has been observed that IL-1Ra injected intra-articularly within 30 days of ACL injury (n=6) reduced pain and improved function at 2 weeks compared to placebo (n=5).90 Although this strategy has proved to be efficacious in the early postinjury phase, the results obtained have not been confirmed in larger studies. Moreover, the ability of IL-1 inhibition to prevent the long-term onset of chronic PTA has not been demonstrated.
Conclusion
PTA frequently develops following joint trauma. Inflammatory events in the initial phase after injury, such as the increased release of inflammatory cytokines, can predispose to the development of OA or inflammatory arthritis.
PTA treatment is a challenging issue to manage. Currently, biochemical markers that predict or correlate with the progression of disease are lacking, and interventions are limited to restoration and stabilisation of the joint. Anti-inflammatory therapy, in particular intra-articular cytokine inhibition, may provide an effective approach for reducing or preventing chronic disability.
Thus, further studies are needed to identify specific markers for the early detection of the disease progression, and to explore innovative opportunities for prevention of future chronic disease.
References
Footnotes
Contributors LP and AS drafted and revised the paper. PG, RL, RR and FO drafted the paper. MF revised the paper.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.
Data sharing statement No additional data are available.