Article Text

Abstract
Objectives Genetic variation in the renal urate transporters SLC2A9 (GLUT9) and SLC22A11 (OAT4) has been reported to interact with diuretics to increase the risk of developing gout. The aim of this study was to determine whether variation in SLC2A9 or SLC22A11 influences acute renal handling of uric acid in response to frusemide.
Methods Following an overnight fast, healthy participants (n=100) attended a study visit with oral intake of 40 mg frusemide. Blood and urine samples were obtained at baseline and 30, 60, 120 and 180 min after frusemide intake. The primary end point was change in fractional excretion of uric acid (FEUA).
Results Following intake of frusemide, FEUA initially increased (mean (SD) change from baseline +1.9% (3.0%) at 60 min, p<0.001) and then decreased (mean (SD) change from baseline −1.5% (2.1%) at 180 min, p<0.001). A very small increase in serum urate was observed over the study period (mean (SD) change from baseline 0.007 (0.01) mmol/L at 180 min, p<0.001). The presence of the urate-lowering and gout-protective alleles for SLC2A9 (rs11942223 and rs13129697) and SLC22A11 (rs207826) did not significantly alter the FEUA following a frusemide load. At both 60 and 180 min, change in fractional excretion of sodium was independently associated with change in FEUA (standardised β≥0.40, p<0.001).
Conclusions The tested variants in SLC2A9 and SLC22A11 do not influence acute changes in renal handling of uric acid in response to frusemide.
Trial registration number ACTRN12614000871640; Results.
- Gout
- Gene Polymorphism
- Pharmacogenetics
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Diuretic use is a risk factor for hyperuricaemia and gout.
A recent population-based study reported that variants in SLC2A9 (encoding GLUT9) and SLC22A11 (encoding OAT4) interact with diuretic use to increase the risk of developing gout.
What does this study add?
This study does not support the hypothesis that genetic variation in SLC2A9 or SLC22A11 influences acute changes in renal handling of uric acid in response to diuretic intake.
How might this impact on clinical practice?
Testing for genetic variants in SLC2A9 or SLC22A11 is unlikely to predict changes in renal handling of uric acid or serum urate in response to diuretic intake.
Hyperuricaemia is frequently observed in patients taking diuretics.1 Short-term intervention studies have shown that oral intake of diuretics reduces uric acid excretion and increases serum urate concentrations.2–4 In addition, diuretic use is a risk factor for the development of incident gout.5 It has been postulated that the association between diuretics and hyperuricaemia is mediated through interactions between diuretics and urate transporters in the renal proximal tubule.6 A variety of urate transporters on the apical and basolateral membranes of the proximal tubule influence uric acid reabsorption from, and secretion into, the tubular lumen to regulate the fractional excretion of uric acid (FEUA) and ultimately determine serum urate concentrations.7 In in vitro assays, diuretics influence the function of a number of these transporters, particularly the organic anion transporters (OATs).8
An analysis of the Atherosclerosis Risk in Communities (ARIC) study reported that specific genetic variants in two urate transporters, specifically SLC2A9 (encoding GLUT9) and SLC22A11 (encoding OAT4), interact with diuretic use to increase the risk of developing gout at a population level.6 Although not consistently observed,9 the ARIC study findings suggest that variation in SLC2A9 and SLC22A11 may influence renal uric acid handling in response to diuretics. Genome-wide association studies (GWAS) of serum urate, FEUA and gout have reported a strong association with common variants of SLC2A9.10 Variation in SLC22A11 has also been associated with hyperuricaemia in a large GWAS of Europeans.10 Several SLC2A9 single nucleotide polymorphisms (SNPs) have been implicated in the development of gout in Māori and Pacific people living in New Zealand,11 who have a very high prevalence of early onset and severe gout.12 ,13 SLC22A11 is also associated with gout in Māori and Pacific people living in New Zealand.14 The functional mechanisms underlying the GWAS-identified association of these variants on urate transport is not currently understood.15
The aim of this study was to examine the influence of SLC2A9 and SLC22A11 in the FEUA following intake of frusemide.
Methods
Participants and methods
This acute intervention study was designed to examine the influence of SLC2A9 and SLC22A11 variation on diuretic-induced renal urate transport. The protocol was modified from previous short-term studies investigating the acute effect of oral frusemide on serum urate, FEUA and allopurinol dosing.2 ,3 ,16 Young healthy participants without kidney disease were recruited into the study to allow the examination of gene–frusemide interactions in the context of normal renal tubular function. The primary end point was change in FEUA over 180 min after frusemide ingestion.
Participants
One hundred healthy participants were recruited by public advertisement. The study protocol prespecified recruitment of 50 participants of Māori or Pacific ancestry and 50 participants of European ancestry. Ancestry was determined by self-report. Inclusion criteria were: age between 18 and 50 years, able to provide written informed consent, and estimated glomerular filtration rate >60 mL/min/1.73 m2. Exclusion criteria were history of gout, history of diabetes, fasting capillary glucose >6 mmol/L, existing diuretic use, hypokalaemia (serum potassium<3.5 mmol/L), recurrent episodes of vasovagal syncope or fainting, and previous intolerance to frusemide.
Potential participants attended a screening visit, during which a general health questionnaire was completed and baseline measurements (including weight, height and waist circumference) and physical examination were performed. Screening blood tests were also carried out. The first participant visit was in September 2014 and the last participant completed the study in November 2015. All visits took place at a clinical research facility in a tertiary medical centre. The study was approved by the New Zealand Ministry of Health Multiregional Ethics Committee (MEC/05/10/130/AM06), and each participant gave written informed consent. The study was registered by the Australian Clinical Trials Registry (ACTRN12614000871640).
Māori and Pacific participants were recruited as a single group of Polynesian ancestry, as the allele frequencies for the SLC2A9 and SLC22A11 variants studied are similar in these two groups,11 ,14 and our previous data have shown similar renal responses to hyperuricaemia in Māori and Pacific participants.17 We included a 1 week period of salt restriction prior to the study visit to standardise the renal urate responses to frusemide and reduce variation due to non-genetic factors.18
Protocol
The study visit occurred within 2 weeks of the screening visit. Following an overnight fast, participants attended a study visit. Online supplementary table S1 shows the study visit timeline. At this visit, participants were given 500 mL water and a light low-purine breakfast at 08:00. A venous catheter was inserted for blood collection. A single oral dose of frusemide 40 mg was given at 09:00, and blood was obtained for urate, sodium, potassium, creatinine and serum storage, prior to frusemide ingestion and then 30, 60, 120 and 180 min after ingestion. Blood was also obtained for genomic DNA extraction at baseline. Urine volume was measured and urine was obtained at each time point for testing of urate, sodium, potassium and creatinine. Water was also provided at each time point (30, 60, 120 and 180 min) to a volume equivalent to the collected urine volume. Additional details about the study protocol are shown in the online supplementary methods.
supplementary tables
supplementary methods
Laboratory testing
Serum and urine chemistry were tested using the Roche Modular P (Hitachi) analyser. Urate concentrations are reported in mmol/L (for conversion to mg/dL, multiply value in mmol/L by 16.81). The FEUA was calculated; this is the ratio between the renal clearance of uric acid to the renal clearance of creatinine, expressed as a percentage. The fractional excretion of sodium (FENa) and fractional excretion of potassium (FEK) were also calculated for each time point. Genotyping of the following SNPs: SLC2A9 rs11942223 and rs13129697, and SLC22A11 SNP rs2078267 was performed on DNA extracted from whole blood samples by a guanidinium-HCl and chloroform extraction-based method. The rs11942223 variant from SLC2A9 was genotyped using a TaqMan SNP genotyping assay (Thermo Fisher Scientific, Assay ID: C_1216479_10) in a Lightcycler 480 Real-Time PCR System (Roche Molecular Systems). Genotypes were autocalled by the Lightcycler 480 software, and the reporter dye plots were visually inspected for correct genotype clustering by a trained analyst. The other two SNPs rs13129697 from SLC2A9 and rs2078267 from SLC22A11, were extracted from whole-genome genotyping performed on the Illumina Infinium CoreExome V.24 bead chip platform at the University of Queensland (Centre for Clinical Genomics). Bead chip genotypes were autoclustered using GenomeStudio V.2011.1 software (Illumina). Clusters were reviewed by a trained analyst following the Illumina GenomeStudio best practice guidelines and quality control protocols of Guo et al19 to ensure final genotype calls were of the highest possible quality.20
Statistical analysis
The primary end point was a comparison of the FEUA response to frusemide based on the presence or absence of the SLC2A9 SNP rs11942223 urate-lowering and gout-protective C allele and independently the presence or absence of the SLC22A11 SNP rs2078267 urate-lowering and gout-protective T allele. The key secondary end points were change in serum urate, and a comparison of the change in FEUA between ancestral subgroups. The study protocol prespecified SLC2A9 rs11942223 as the SLC2A9 SNP for the primary analysis; this SNP was selected due to the strong association with hyperuricaemia and gout in European and Polynesian populations.10 ,11 ,21 In addition, the SLC2A9 SNP rs13129697 was included in further exploratory analyses, as rs13129697 was reported in the ARIC study that described the interaction between SLC2A9 and diuretic use in risk of incident gout in European participants.6 Linkage disequilibrium (r2) between rs11942223 and rs13129697 was 89% in European participants and 26% in Māori and Pacific participants.
Sample size calculations were based on the previous study by Yu et al.3 Using this estimate of variability, a sample size of 100 participants had 90% power at the α=0.05 significance level to detect a biologically relevant absolute mean difference of at least 1 FEUA unit between those with (n=30) and without (n=70) the protective SLC2A9 rs11942223 allele,11 and between those with (n=50) and without (n=50) the protective SLC22A11 rs2078267 allele,14 3 hours after frusemide (the primary end point). See online supplementary methods for further details.
Analyses were performed using SAS (SAS Institute V.9.2) on an intention-to-treat basis. Data are presented as mean (SD) or n (%) for descriptive purposes; however, measures of effect are presented with the appropriate 95% CI. A mixed-model approach to repeated measures (analysis of covariance, ANCOVA) was employed, modelling the change from baseline in FEUA as the dependent variable and including baseline FEUA as a covariate. Time, the presence or absence of the protective allele and their interaction was modelled. Unstructured, compound symmetry and first-order autoregressive covariance structures were examined and unstructured, which had the smallest Akaike information criterion, was subsequently used in all analyses. Significant main and/or interaction effects were explored using the method of Tukey to preserve the overall 5% significance level within each analysis. Age, sex and ancestry were adjusted for within the models. Prespecified comparison of change from baseline between alleles at 30, 60, 120 and 180 min was made using error terms calculated from the mixed model, an overall 5% significance level was maintained using a false discovery rate protected p value for these comparisons. Stepwise linear regression was used to identify variables that were independently associated with changes in FEUA and serum urate. See online supplementary methods for further details.
Results
Participants
There were 131 prospective participants screened. Thirty-one prospective participants were excluded following the screening visit for the following reasons: 14 had serum potassium <3.5 mmol/L, 12 did not meet ancestry criteria, 2 had a history of gout, 2 had fasting capillary glucose >6 mmol/L and 1 had veins unsuitable for cannulation. All other participants (n=100) attended the study visit.
Characteristics of participants attending the study visit at baseline are shown in table 1. The ancestral groups were generally well matched, although Māori or Pacific participants were older, and had higher baseline serum urate and lower baseline serum potassium. The protective SLC22A11 SNP rs2078267T allele was less frequent in the Māori or Pacific group.
Characteristics of participants at baseline
Adverse events during the study visit were noted in four participants; two participants experienced hypotension and nausea which resolved following intravenous infusion of 500 mL 0.9% NaCl, one participant experienced severe nausea and bradycardia during a blood draw which resolved by supine positioning, and one participant experienced residual arm pain following venesection. All participants (n=100) completed the study visit.
The effects of frusemide in the entire group
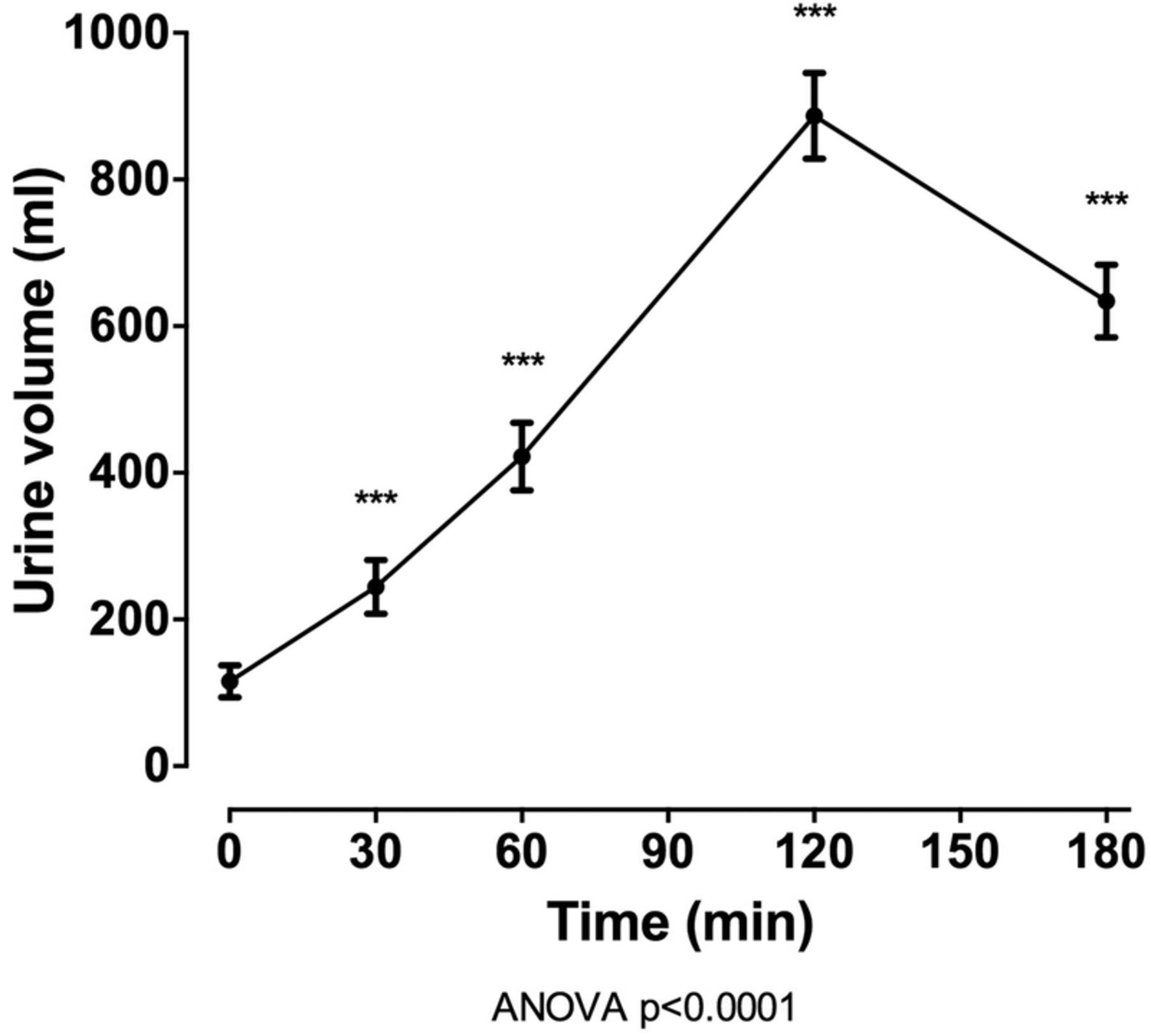
In the entire group (n=100), oral intake of 40 mg frusemide led to a marked diuresis, with mean (SD) urine volume 2161.9 (717.37) mL over 180 min (figure 1). Following intake of frusemide, FEUA initially increased (mean (SD) change from baseline +1.9% (3.0%) at 60 min, p<0.001) and then decreased (mean (SD) change from baseline −1.5% (2.1%) at 180 min, p<0.001; figure 2). A very small increase in serum urate was observed over the study period (mean (SD) change from baseline 0.007 (0.01) mmol/L at 180 min, p<0.001). There was also an increase in FENa and FEK over the 180 min study period (p<0.0001 for all), peaking 120 min after frusemide intake. There was a small increase in serum sodium and potassium over the study period, peaking at 60 min post frusemide intake.
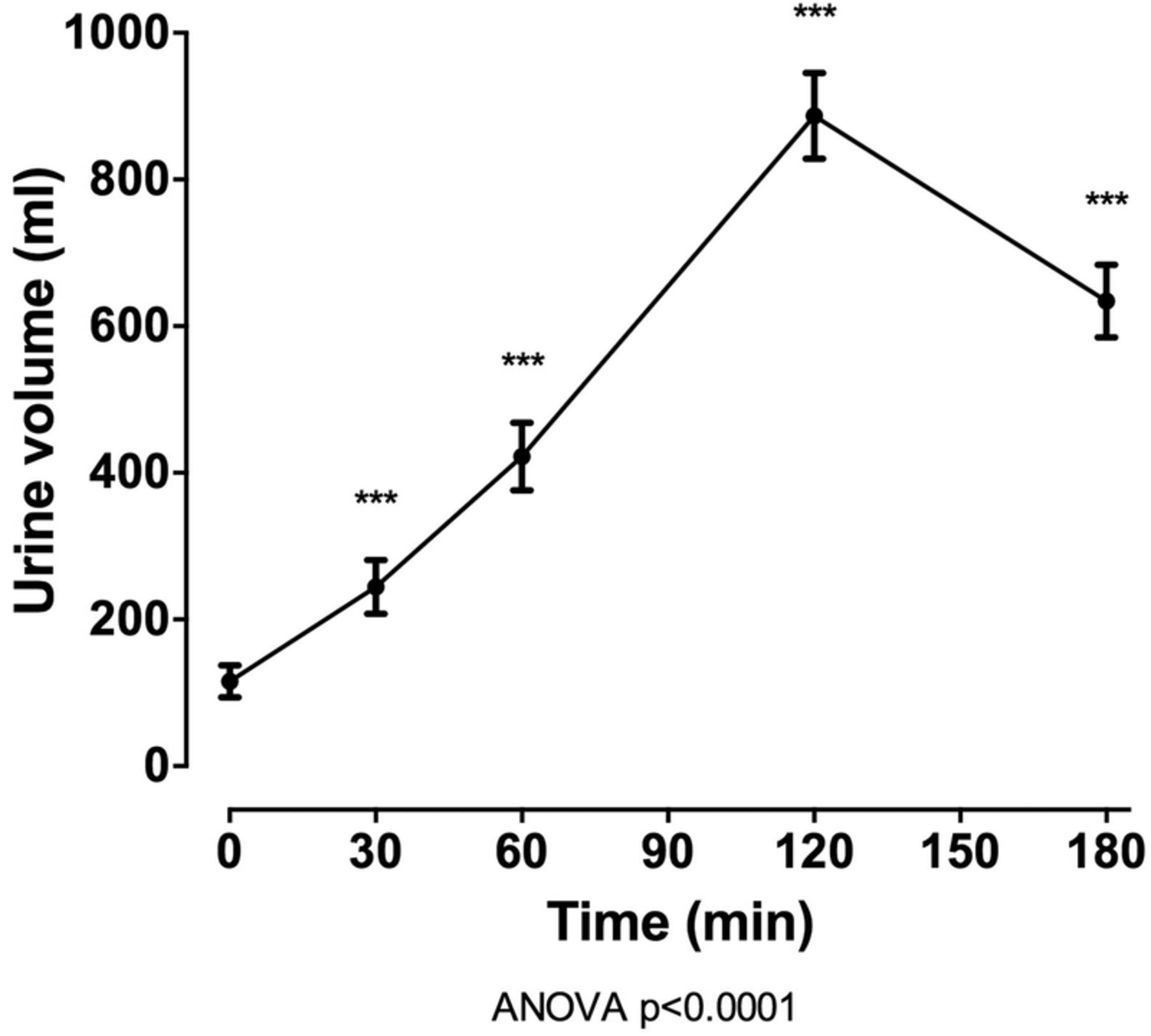
Urine volume over the 180 min study period in the entire group (n=100). Data are presented as mean (95% CI). ANOVA, analysis of variance.

FE and serum concentrations over the 180 min study period for the entire group (n=100). Data are presented as mean (95% CI). Top panels show urate, middle panels show sodium and bottom panels show potassium. ANOVA, analysis of variance; FE, fractional excretion.
Co-primary end point: SLC2A9
In the entire group, the presence of the SLC2A9 rs11942223 protective C allele did not significantly influence changes in FEUA or serum urate following the frusemide load (ANCOVA pSNP≥0.23, figure 3). At baseline and throughout the study period, the serum urate concentration was higher in those without the rs11942223 protective allele (false discovery rate p≤0.013 for all time points, figure 3). However, the changes in FEUA and serum urate over the study period were similar in those with and without the rs11942223 protective allele. Similarly, in the additional SLC2A9 rs13129697 exploratory analysis, the presence of the protective G allele did not influence change in FEUA following the frusemide load (ANCOVA pSNP=0.54, see online supplementary figure S1). Furthermore, there was a greater increase in serum urate in those with the rs13129697 protective allele present (ANCOVA pSNP=0.03), but no difference in the absolute values in serum urate over the study period (false discovery rate p>0.49 for all time points, see online supplementary figure S1).
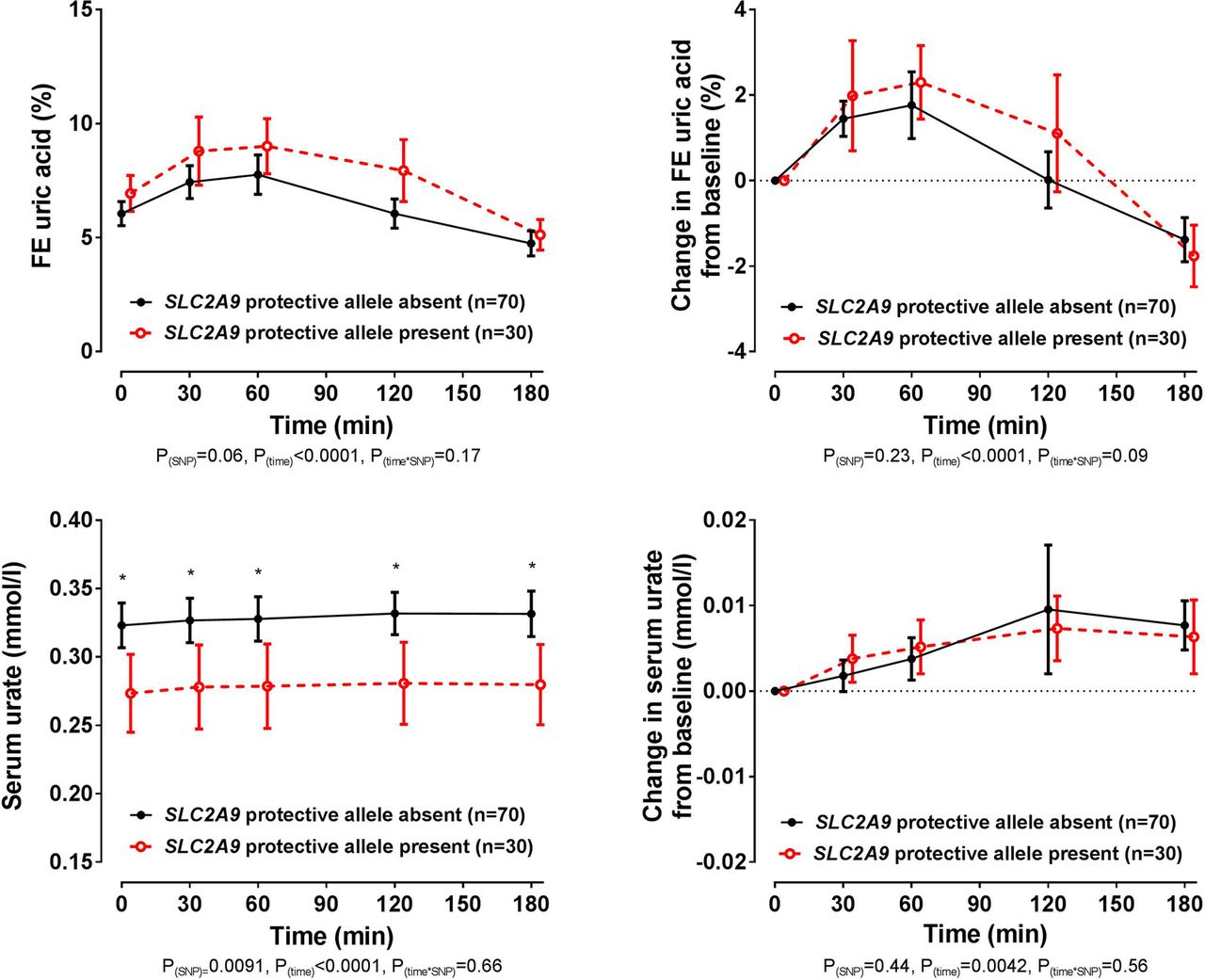
Fractional excretion (FE) of uric acid (FEUA) and serum urate concentrations over the study period, based on the presence or absence of the SLC2A9 rs11942223 protective C allele. Data are presented as mean (95% CI). Left panel shows absolute values and right panel shows change in values. Age-adjusted, sex-adjusted and ancestry-adjusted p values are shown. *False discovery rate p<0.05, for comparison between SLC2A9 rs11942223 allele groups at the specified time point.
supplementary figure
Co-primary end point: SLC22A11
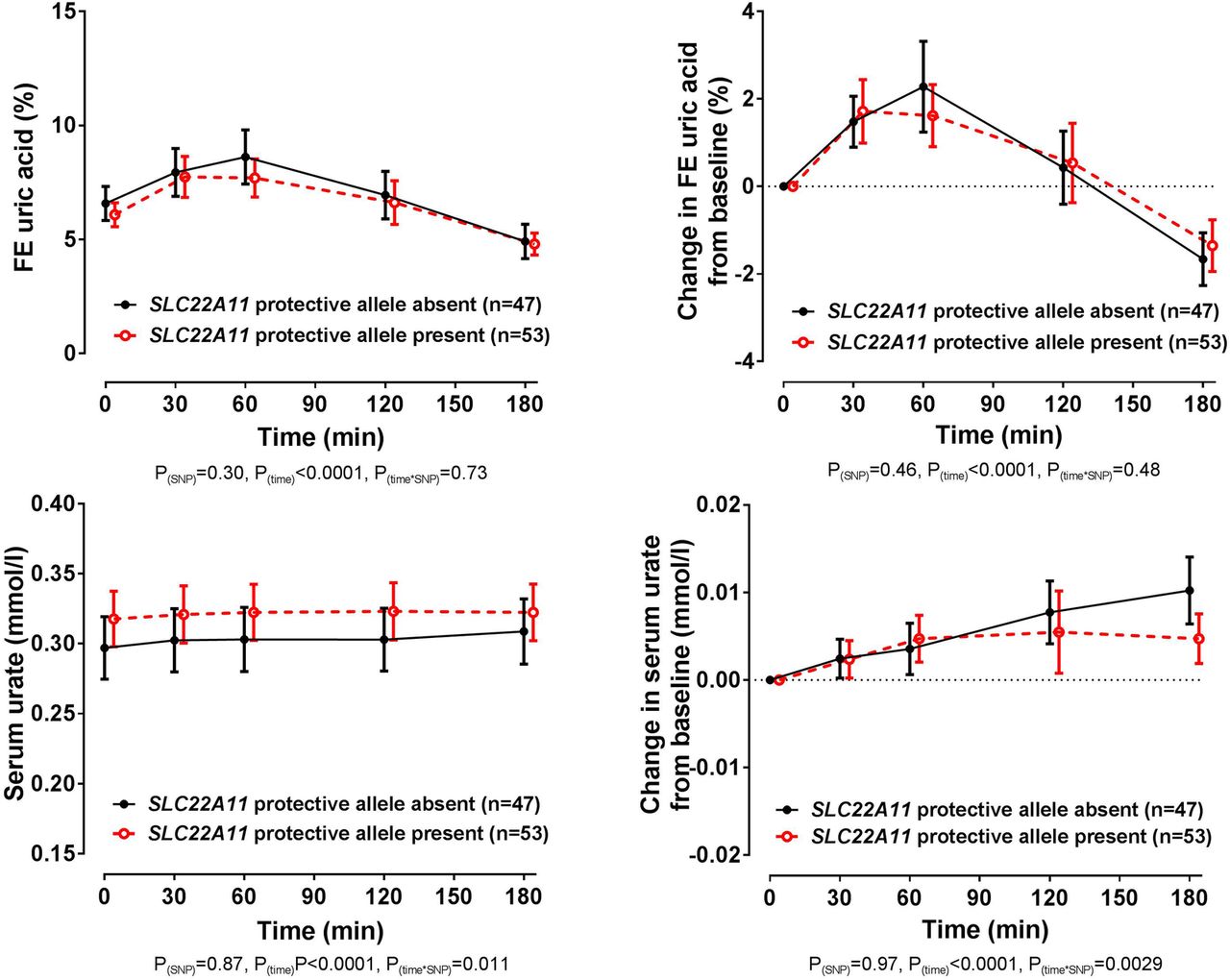
In the entire group, there was no significant difference in baseline FEUA or serum urate in those with and without the SLC22A11 rs2078267 protective T allele (false discovery rate p>0.58). Furthermore, the changes in FEUA over the study period were similar in those with and without the rs2078267 protective allele (ANCOVA pSNP=0.46, figure 4). Despite the lack of effect on FEUA, there was a difference in the direction of the change in serum urate depending on rs2078267 genotype, with flattening of the serum urate increase in those with the protective T allele (ANCOVA ptime×SNP=0.0029, figure 4). However, the change in serum urate from baseline was very small, and absolute serum urate concentrations did not differ in the SLC22A11 groups at the end of the study period (false discovery rate p>0.98 at the 180 min time point, figure 4).
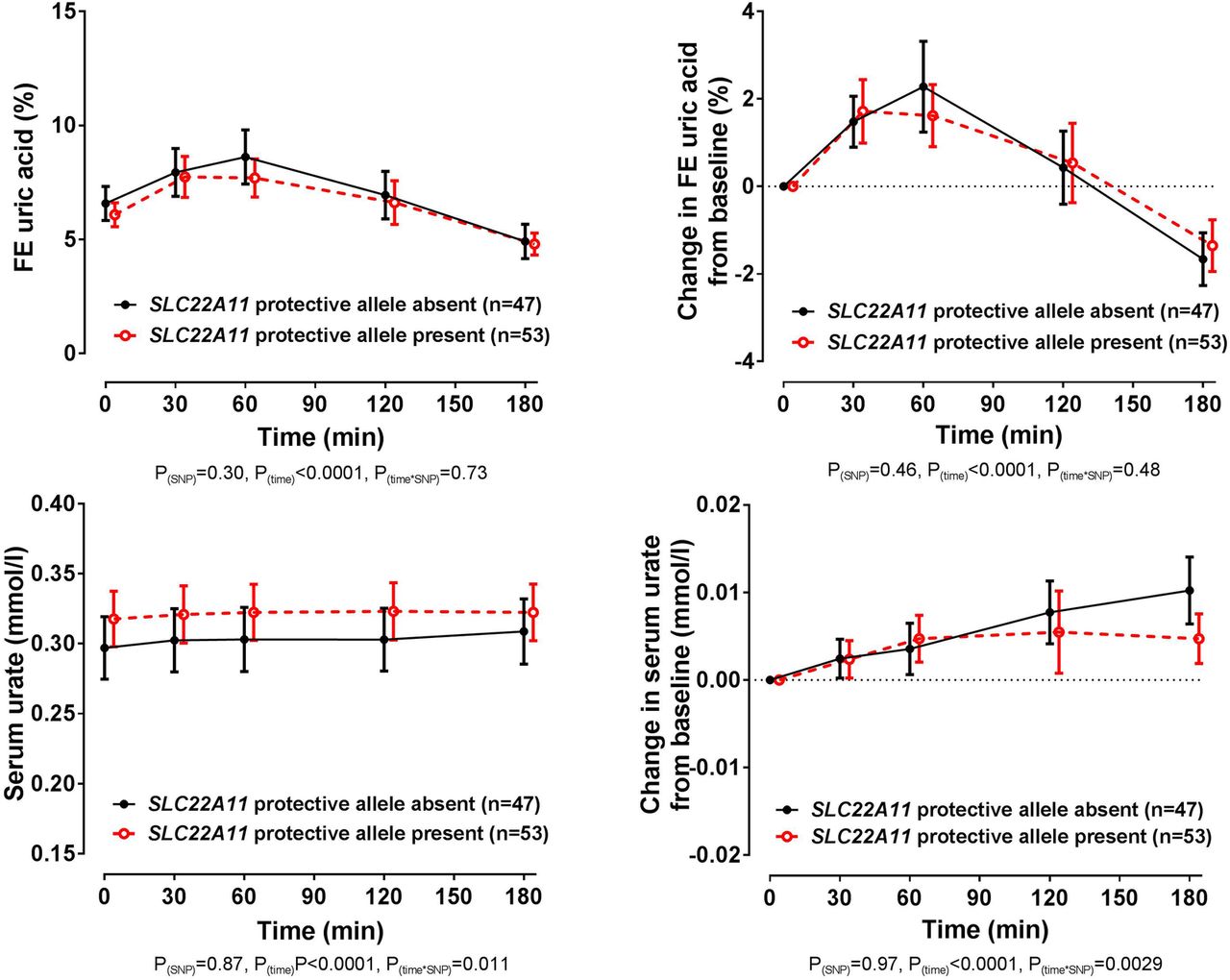
Fractional excretion (FE) of uric acid (FEUA) and serum urate concentrations over the study period, based on the presence or absence of the SLC22A11 rs2078267 protective T allele. Data are presented as mean (95% CI). Left panel shows absolute values and right panel shows change in values. Age-adjusted, sex-adjusted and ancestry-adjusted p values are shown.
Ancestral stratification
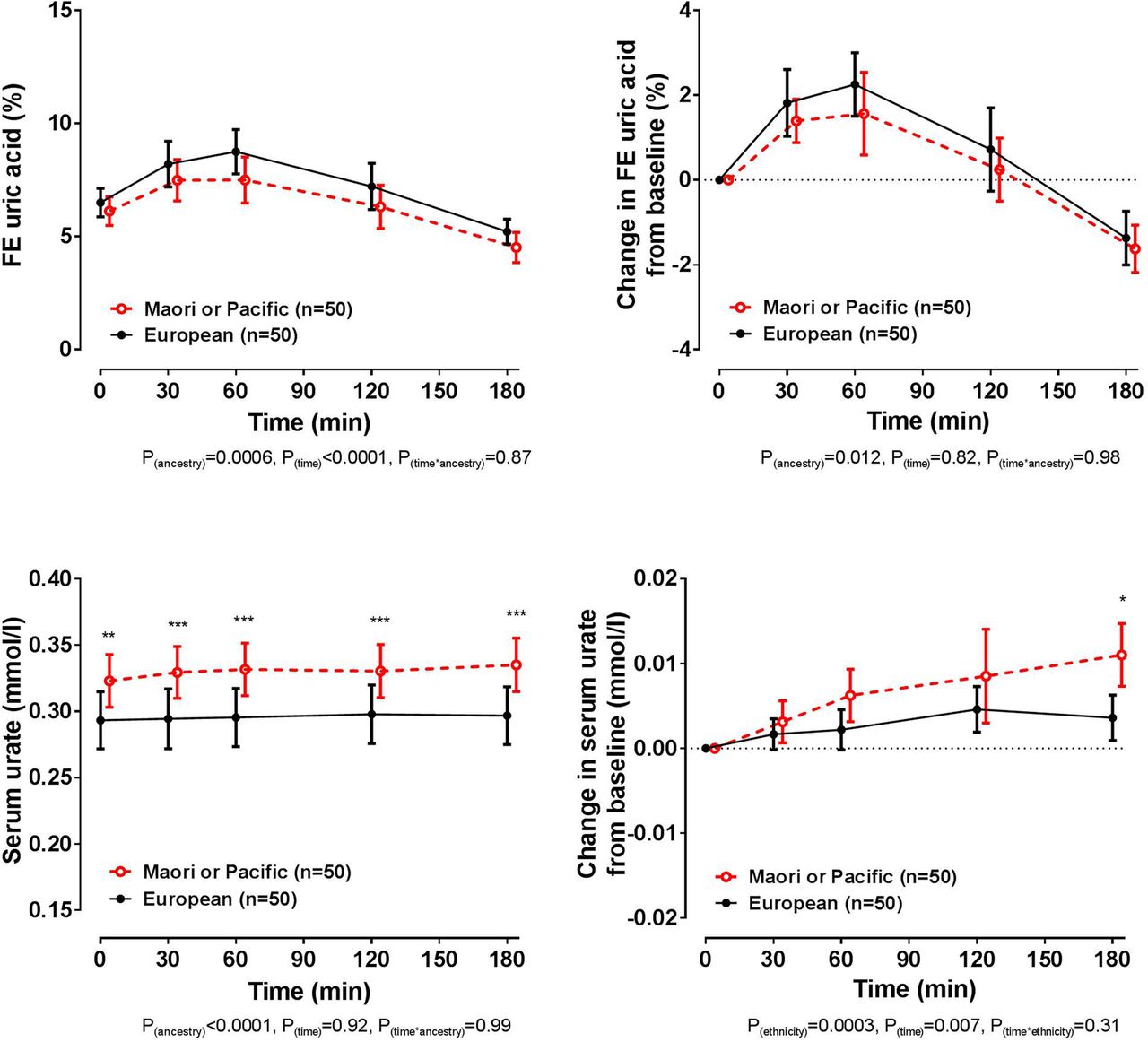
At baseline, the European and Polynesian ancestral groups had similar FEUA (false discovery rate p=0.37, figure 5). The biphasic FEUA response to frusemide was observed in both ancestral groups, although the Māori or Pacific group had a smaller increase in FEUA following intake of frusemide overall (ANCOVA pancestry=0.012). At baseline, the serum urate concentration was higher in those of Māori or Pacific ancestry (false discovery rate p=0.001). Change in serum urate from baseline was small in both ancestral groups, although the Māori or Pacific group had a greater increase in serum urate concentration over the study period (ANCOVA pancestry=0.0003), with a significantly greater increase between ancestral groups at the 180 min time point (false discovery rate p=0.011). The presence of the protective alleles for SLC2A9 or SLC22A11 did not significantly alter the FEUA or serum urate responses to the frusemide load in either ancestral group (ANCOVA pSNP≥0.10 for all comparisons, see online supplementary tables S2–S4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Fractional excretion (FE) of uric acid (FEUA) and serum urate concentrations over the study period, based on ancestral group. Data are presented as mean (95% CI). Left panel shows absolute values and right panel shows change in values. Age-adjusted and sex-adjusted p values are shown. *False discovery rate p<0.05, **false discovery rate p<0.01 and ***false discovery rate p<0.001, for comparison between ancestral groups at the specified time point.
Predictors of change in FEUA and serum urate
At the 60 min time point, change in FEUA was independently associated with change in FENa (standardised β=0.40, p<0.001, see online supplementary tables S5 and S6). Similarly, at the 60 min time point, change in serum urate was independently associated with change in FENa (standardised β=0.44, p<0.001). At the 180 min time point, change in FEUA was also independently associated with change in FENa (standardised β=0.46, p<0.001), and change in serum urate was positively associated with change in serum urate at 60 min (standardised β=0.59, p<0.001) and inversely associated with change in FEUA at 180 min (standardised β=−0.21, p=0.009). Urine volume was not independently associated with change in FEUA or change in serum urate at either time point.
Discussion
This short-term intervention study does not support the hypothesis that genetic variation in SLC2A9 or SLC22A11 influences acute changes in renal handling of uric acid in response to diuretic intake. It is conceivable that genetic variants in other urate transporters (either renal or extrarenal) not tested in this analysis do influence these responses. However, in the ARIC population-based study, the effects of genetic risk scores (that included variants in the ABCG2 and SLC17A1 uric acid transporter genes in addition to SLC2A9 and SLC22A11) on diuretic-associated gout were driven by SLC2A9 and SLC22A11 variants.6 The conclusions of our analysis are consistent with another population-based study that failed to replicate the ARIC study findings.9
The pattern of FEUA changes over time following intake of oral frusemide was biphasic, with initial increase and subsequent reduction in FEUA. The initial increase in FEUA does not explain the observed increase in serum urate over the study period, suggesting that acute serum urate-increasing effects of diuretics are not directly related to interactions with urate transporters. The changes in FEUA were independently associated with changes in FENa. A number of urate transporters, including URAT1, OAT1, OAT, and OAT10 are influenced by substrates transported by sodium-dependent anion co-transporters,7 and our data support the concept that diuretics may influence renal uric acid handling indirectly, through effects on sodium transport.
Our results showing the biphasic FEUA response differ from prior analysis of oral frusemide dosing4 but are similar to some studies of intravenous frusemide, which also showed a similar initial increase, then reduction, in FEUA.18 ,22 In the Steele and Oppenheimer's18 study of intravenous frusemide, late reduction in FEUA was attenuated by volume repletion, suggesting that the effects of frusemide on tubular function were related to volume depletion. In our analysis, changes in FEUA at 60 min and 180 min after frusemide were not independently associated with urine volume, but rather with changes in FENa. The relationship between renal clearance of uric acid and FENa has also been reported in patients with gout.23 Collectively, these data suggest an interplay between uric acid and sodium clearance.
Māori and Pacific people have very high rates of hyperuricaemia and early-onset gout,12 with reported differences in renal handling of urate.24 ,25 Consistent with a previous short-term intervention study of fructose intake,17 we observed differences between ancestral groups in this short-term intervention study of frusemide, with higher baseline serum urate, lower increase in FEUA and greater increase in serum urate over the 180 min follow-up period in renal urate handling in Māori and Pacific participants, compared with New Zealand European participants. However, these differences were not due to genetic variants tested in SLC2A9 or SLC22A11.
This study has some limitations; the study was designed to examine the effects of SLC2A9 or SLC22A11, based on prior report of these genes contributing to diuretic-associated gout. We cannot exclude that other variants in SLC2A9 and SLC22A11 or in other genes (urate, sodium or potassium transporters) may influence renal responses to frusemide. A validation study would be of great interest, to confirm the observed lack of effect of the tested variants. The observed acute serum urate increase in response to a single frusemide in these healthy volunteers was very small and the clinical relevance of these findings is uncertain. The study was designed to recruit participants with normal tubular function, and most participants lacked other factors associated with diuretic use such as kidney or cardiovascular disease; it is possible that these comorbidities interact with genetic variants and/or diuretics to influence FEUA, serum urate or gout risk. This study was designed to examine the short-term effects of frusemide only; long-term administration may have different effects on FEUA or serum urate.3
Nevertheless, these data do not support the hypothesis that genetic variation in SLC2A9 or SLC22A11 plays a role in diuretic-associated gout. Testing for these variants is unlikely to predict changes in renal handling of uric acid or serum urate in response to diuretic intake.
References
Footnotes
Contributors ND (the guarantor) accepts full responsibility for the work and the conduct of the study, had access to the data and controlled the decision to publish. ND conceived the study, contributed to the data interpretation, and drafted the manuscript. JA and AH recruited participants, coordinated study visits and contributed to data acquisition. JA and BM managed clinical data management, AP-G and TJF contributed to data acquisition and genetic data management. GDG analysed the data. RD, LKS and TRM conceived of the study, contributed to the data interpretation and drafted the manuscript. All authors read and approved the final manuscript.
Funding This work was supported by the Health Research Council of New Zealand (grant number 14-527).
Competing interests ND reports grant funding, consultancy or speaker fees from Takeda, Teijin, Menarini, Pfizer, Ardea Biosciences/AstraZeneca, Cymabay and Crealta, all outside the submitted work. LKS reports grant funding and consultancy from Ardea Biosciences/AstraZeneca, all outside the submitted work. TRM reports grant funding and consultancy from Ardea Biosciences/AstraZeneca, all outside the submitted work.
Ethics approval New Zealand Ministry of Health Multiregional Ethics Committee (MEC/05/10/130/AM06).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.