Article Text

Abstract
Objective Tumour necrosis factor alpha (TNF-α) plays an important role in rheumatoid arthritis (RA). TNF-α is synthesised as a membrane-anchored precursor and is fully activated by a disintegrin and metalloproteinase 17 (ADAM17)-mediated ectodomain shedding. Nardilysin (NRDC) facilitates ectodomain shedding via activation of ADAM17. This study was undertaken to elucidate the role of NRDC in RA.
Methods NRDC-deficient (Nrdc–/– ) mice and macrophage-specific NRDC-deficient (NrdcdelM ) mice were examined in murine RA models, collagen antibody-induced arthritis (CAIA) and K/BxN serum transfer arthritis (K/BxN STA). We evaluated the effect of gene deletion or silencing of Nrdc on ectodomain shedding of TNF-α in macrophages or monocytes. NRDC concentration in synovial fluid from patients with RA and osteoarthritis (OA) were measured. We also examined whether local gene silencing of Nrdc ameliorated CAIA.
Results CAIA and K/BxN STA were significantly attenuated in Nrdc–/– mice and NrdcdelM mice. Gene deletion or silencing of Nrdc in macrophages or THP-1 cells resulted in the reduction of TNF-α shedding. The level of NRDC is higher in synovial fluid from RA patients compared with that from OA patients. Intra-articular injection of anti-Nrdcsmall interfering RNA ameliorated CAIA.
Conclusion These data indicate that NRDC plays crucial roles in the pathogenesis of autoimmune arthritis and could be a new therapeutic target for RA treatment.
- TNF-alpha
- Rheumatoid arthritis
- Synovial fluid
- Autoantibodies
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Tumour necrosis factor alpha (TNF-α), which is activated by ectodomain shedding, plays an important role in rheumatoid arthritis (RA).
Nardilysin facilitates TNF-α shedding via enhancing a disintegrin and metalloproteinase 17 activity.
What does this study add?
Deletion or inhibition of nardilysin prevents autoantibody-induced arthritis, murine models of RA.
Concentration of nardilysin in synovial fluid is markedly increased in patients with RA compared with that in patients with osteoarthritis, suggesting a crucial role of nardilysin in the pathogenesis of RA.
How might this impact on clinical practice?
Nardilysin could be a novel biomarker and therapeutic target for RA.
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease that causes joint inflammation and destruction affecting 0.5%–1.0% of the population.1 Anti-tumour necrosis factor alpha (TNF-α) biologic agents have led to a revolutionary improvement in the management of RA patients, indicating that this inflammatory cytokine plays a key role in RA pathogenesis. The importance of TNF-α has been also demonstrated in murine models of RA; for example, human TNF-α transgenic mice develop arthritis,2 TNF-α is produced in synovial tissues of murine experimental models of RA,3 murine experimental arthritis is attenuated in TNF-α-deficient mice4 and neutralisation of TNF-α ameliorates murine collagen-induced arthritis.5 TNF-α is produced mostly by macrophages in RA synovial tissues6 and, alone or in combination with other cytokines or chemokines, recruits inflammatory cells such as B cells, T cells and neutrophils, and induces production of cytokines, chemokines, matrix metalloproteinases or receptor activator of nuclear factor κB ligand, resulting in joint inflammation and bone resorption.7–11
One of the earliest subclinical features of RA is a breakdown of self-tolerance. Autoantibodies such as rheumatoid factor (RF) and anti-citrullinated peptide antibody (ACPA) are found in the patients’ serum long before arthritis occurs.12 While the autoantigens have not been fully identified, autoantibodies may contribute to the pathogenesis of RA via immunocomplex (IC) formation and deposition in synovial tissues.13 The importance of autoantibodies has been demonstrated in mouse autoantibody-induced arthritis (AAIA) models of RA, such as collagen antibody-induced arthritis (CAIA) and K/BxN serum transfer arthritis (K/BxN STA). Antibodies against intra-articular antigens, type II collagen in CAIA14 15 and glucose-6-phophoisomerase in K/BxN STA16 17 are arthrogenic in mice. TNF-α has been demonstrated to be important in both models: anti-TNF-α antibody attenuates CAIA,18 while TNF-α-deficient mice are resistant to K/BxN STA.4
Importantly, TNF-α is synthesised as a membrane-anchored precursor and is fully activated by a disintegrin and metalloproteinase 17 (ADAM17)-mediated ectodomain shedding,19 indicating that ADAM17 is an attractive alternative target for treatment of RA. However, the multiple disease phenotypes of ADAM17-deficient patients and mice have raised concerns about the potential adverse effects of anti-ADAM17 therapy.20 21 We have previously shown that ADAM17 activity and TNF-α shedding are enhanced by nardilysin, N-arginine dibasic convertase (NRDC), via its direct binding to ADAM17.22–24
NRDC is a zinc peptidase of the M16 family that selectively cleaves dibasic sites.25 26 We identified NRDC on the cell surface as a specific binding partner of heparin-binding epidermal growth factor (EGF)-like growth factor (HB-EGF) and demonstrated that NRDC enhances ectodomain shedding of HB-EGF.27 Our subsequent studies revealed that NRDC enhances ectodomain shedding of not only HB-EGF but also a wide range of other membrane proteins including TNF-α,23 28 neuregulin 129 and amyloid precursor protein.22 We have also demonstrated that NRDC-induced ectodomain shedding plays critical roles in myelination, gastric cancer and Alzheimer’s disease.22–24 28–31
We demonstrate here that NRDC-deficient (Nrdc–/– ) mice and macrophage-specific NRDC-deficient (NrdcdelM ) mice show attenuation of AAIA. We also demonstrate that gene deletion or silencing of Nrdc in mouse macrophages or human monocytic cells results in reduction of TNF-α shedding. Importantly, the level of NRDC is increased in synovial fluid from RA patients compared with that from osteoarthritis (OA) patients. Moreover, intra-articular injection of anti-Nrdc small interfering RNA (siRNA) ameliorates CAIA. Together, these data suggest that NRDC controls AAIA via the regulation of TNF-α shedding and could be a new therapeutic target for RA treatment.
Materials and methods
The full methods are provided in the online supplementary material.
Blood and synovial fluid collection
Synovial fluid was obtained during joint surgery from 20 patients with RA and 17 patients with OA, who had been admitted to Kyoto University Hospital. All of the RA patients met 2010 American College of Rheumatology and European League against Rheumatism (ACR/EULAR) classification criteria for RA. Peripheral blood samples were obtained and the level of C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), RF, and ACPA was examined before surgery. Disease activity score 28 (DAS28) was calculated according to the results of physical examinations and blood tests.32 The full methods are provided in the online supplementary material.
Results
Nrdc-deficient mice are resistant to CAIA
To explore the role of NRDC in RA, we compared wild-type (WT) and Nrdc–/– mice with a BALB/c background using the CAIA model. After the intraperitoneal administration of a five-clone cocktail of monoclonal anti-type II collagen antibodies, we assessed the severity of arthritis daily for 2 weeks. The onset of arthritis was delayed (on day 3 in WT mice and on day 6 in Nrdc–/– mice, respectively) and the average of arthritis score was significantly and markedly lower in Nrdc–/– mice (figure 1A,B). Ankle thickness was also significantly less in Nrdc–/– mice from days 4 to 9 compared with that in WT mice. Histological examination of the ankle joints demonstrated that synovial hypertrophy with infiltration of inflammatory cells and bone and cartilage erosion were reduced or absent in Nrdc–/– mice (figure 1C). These results suggest that NRDC is crucial for AAIA development and progression.

CAIA is attenuated in Nrdc – / – mice. (A) Photographs of the hind paws 4 days and 7 days after the induction of CAIA. Upper panels, wild-type (WT); lower panels, Nrdc – / – mice. (B) Arthritis score (upper) and ankle thickness (lower) were measured daily after the induction of CAIA. n=5 per genotype (mean ± SEM). *p<0.05; **p<0.01, Welch’s t-test. (C) H&E staining of the ankle joints 14 days after the induction of CAIA in WT (upper panels) and Nrdc – / – (lower panels) mice. Scale bars: 500 µm in the 40× magnification and 200 µm in the 100× magnification. Symbols in the 100x magnification images: arrow, bone erosion; arrow head, cartilage erosion; *, hypertrophic synovium.
Macrophage-specific Nrdc-deficient mice are resistant to CAIA
Because macrophages play an important role in the pathogenesis of RA, we next generated NrdcdelM mice by crossing Nrdcflox/flox mice with Lyz2-Cre mice, in which Cre recombinase is expressed under the control of the lysozyme 2 gene (Lyz2) promoter. Because the Nrdcflox/flox and NrdcdelM mice have a C57BL/6 genetic background, which is more resistant to CAIA than BALB/c, we administered 2.5 times more antibody to Nrdcflox/flox and NrdcdelM mice for the CAIA experiments compared with the amount that was used for the Nrdc–/– mice. The results showed that the onset of arthritis was delayed and the severity of arthritis was markedly reduced in NrdcdelM mice compared with controls (figure 2A, B). As was the case for Nrdc–/– mice, synovial hypertrophy and bone erosion were attenuated in NrdcdelM mice (figure 2C). These findings demonstrated that macrophage-specific deletion of NRDC almost completely recapitulated the resistance of Nrdc–/– mice to CAIA.
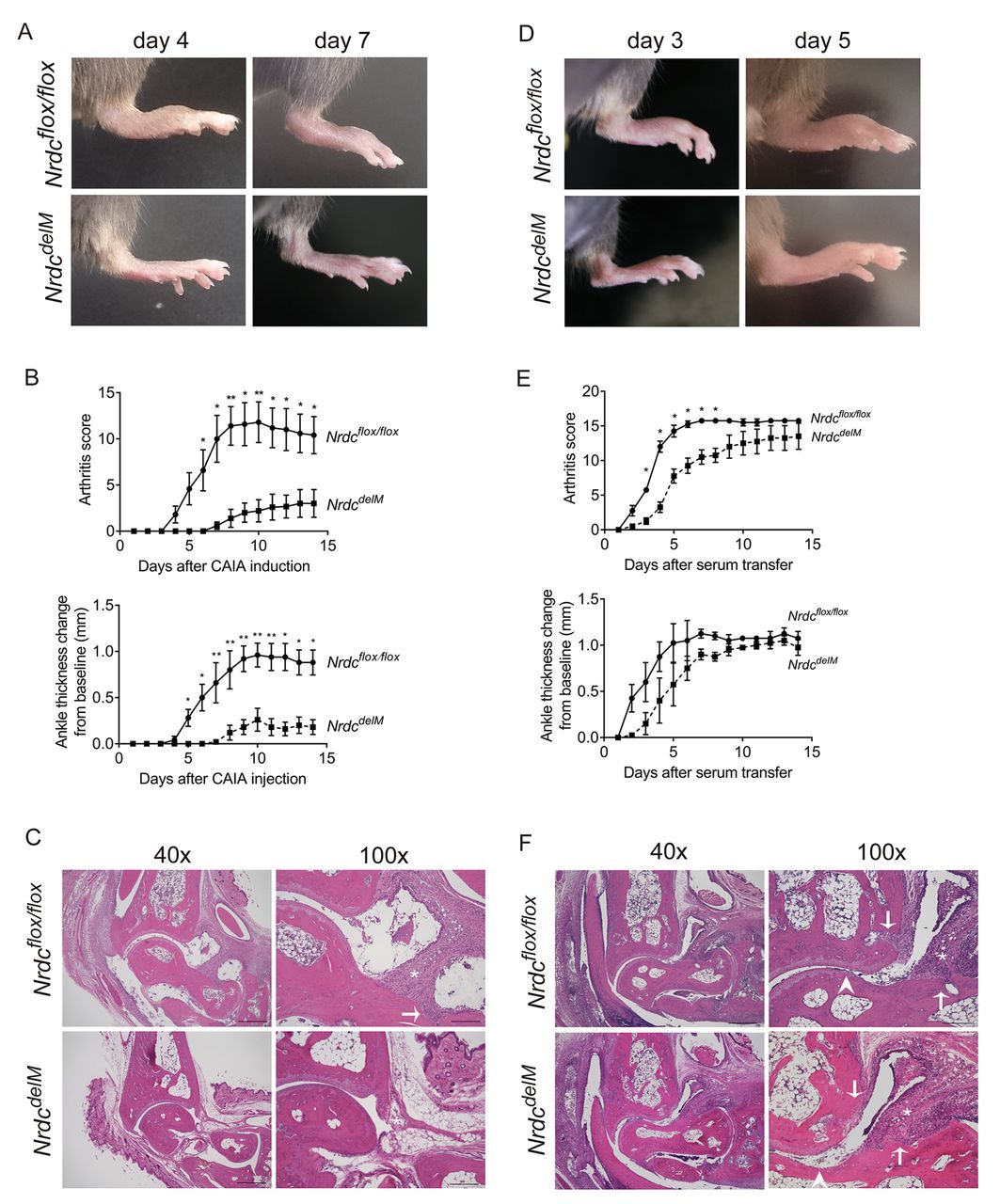
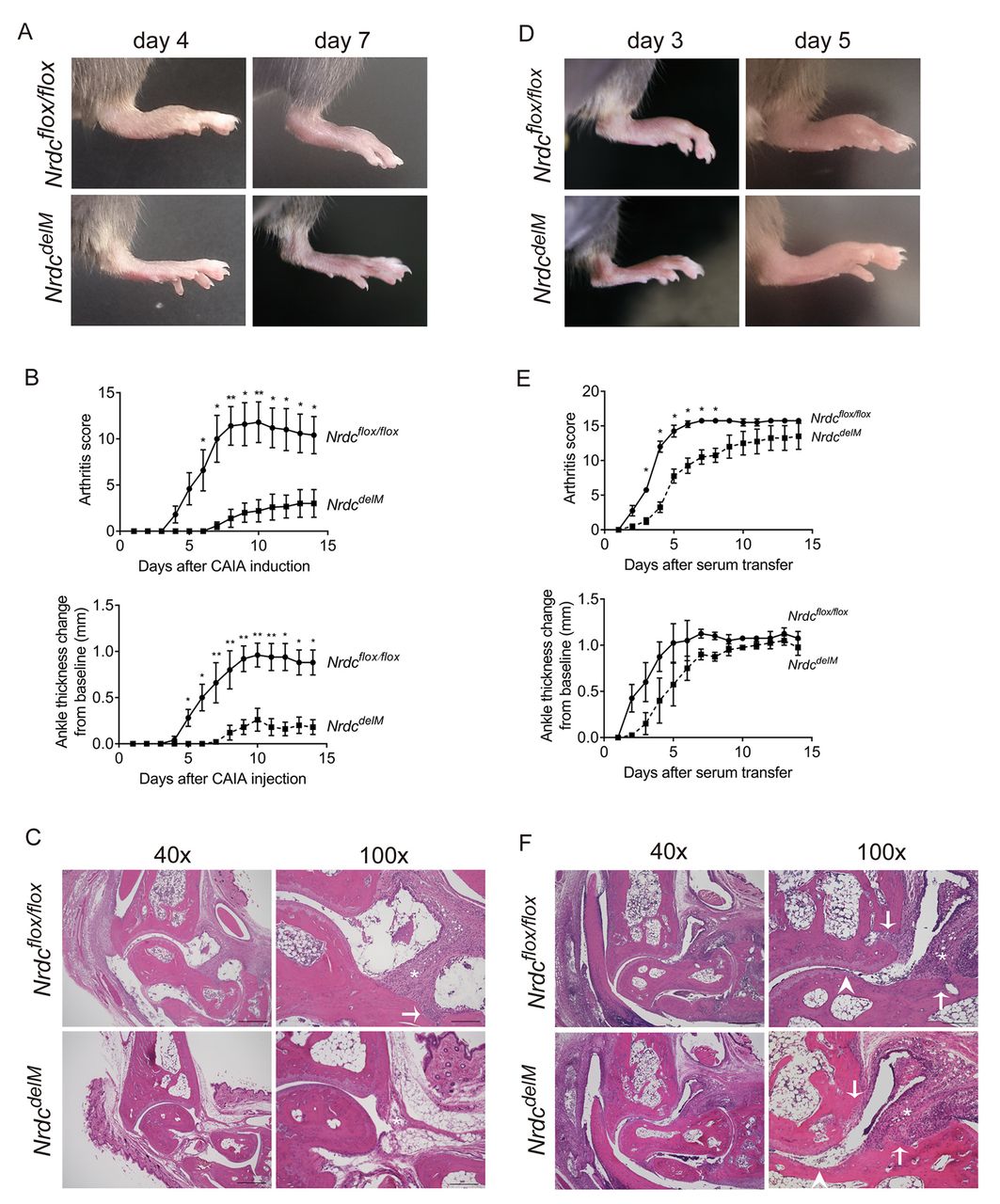
Macrophage-specific Nrdc-deficient (NrdcdelM ) mice are resistant to CAIA and K/BxN STA. (A) Photographs of the hind paws 4 days and 7 days after the induction of CAIA. Upper panels, Nrdcflox/flox ; lower panels, NrdcdelM mice. (B) Arthritis score (upper) and ankle thickness (lower) were measured daily after the induction of CAIA. n=5 per genotype (mean ± SEM). *p<0.05; **p<0.01, Welch’s t-test. (C) H&E staining of the ankle joints 14 days after the induction of CAIA in Nrdcflox/flox (upper panels) and NrdcdelM (lower panels) mice. Scale bars: 500 µm in the 40× magnification and 200 µm in the 100× magnification. Symbols in the 100x magnification images: arrow, bone erosion; *, synovial hypertrophy. (D) Photographs of the hind paws 3 days and 5 days after the induction of K/BxN STA. Upper panels, Nrdcflox/flox ; lower panels, NrdcdelM mice. (E) Arthritis score (upper) and ankle thickness (lower) were measured daily after the induction of K/BxN STA. n=4 per genotype (mean ± SEM). *p<0.05; **p<0.01, Welch’s t-test. (F) H&E staining of the ankle joints 14 days after the induction of K/BxN STA in Nrdcflox/flox (upper panels) and NrdcdelM (lower panels) mice. Scale bars: 500 µm in the 40× magnification and 200 µm in the 100× magnification. Symbols in the 100× magnification images: arrow, bone erosion; arrow head, cartilage erosion; *, synovial hypertrophy.
Arthritis in the K/BxN STA model is attenuated in NrdcdelM mice
To confirm the role of NRDC in autoimmune arthritis, we examined Nrdcflox/flox and NrdcdelM mice in another murine model, the K/BxN STA model. K/BxN mouse serum has been reported to strongly induce arthritis in a wide range of mouse strains.33 As shown in figure 2D,E, K/BxN STA was significantly attenuated in NrdcdelM mice up to day 8 after injection. In contrast, there were no significant differences between Nrdcflox/flox and NrdcdelM mice in the arthritis score after day 9 or in ankle thickness, although both values tended to be lower in NrdcdelM mice (figure 2D,E). The histology of the hind paws at day 14 demonstrated that the synovial hypertrophy with infiltration of inflammatory cells, bone erosion and cartilage erosion were equivalently observed in both Nrdcflox/flox mice and NrdcdelM mice (figure 2F), which is consistent with the results of arthritis score and ankle thickness. Collectively, these data demonstrated that NRDC deficiency in macrophages attenuated the severity of arthritis in two different experimental models of AAIA.
Secretion of TNF-α from macrophages and monocytic cells is reduced by the deletion of Nrdc
Because macrophages are a major source of TNF-α in AAIA34 and NRDC facilitates ectodomain shedding of TNF-α,23 we hypothesised that the attenuation of AAIA in Nrdc–/– mice and NrdcdelM mice was due to the impairment of ectodomain shedding of TNF-α in macrophages. To test this hypothesis, we first examined TNF-α secretion from NRDC-deficient macrophages. We harvested bone marrow derived macrophages (BMDMs) from Nrdcflox/flox and NrdcdelM mice and confirmed that NRDC protein expression was remarkably reduced in BMDMs from NrdcdelM mice (figure 3A). BMDMs were stimulated with or without a low concentration (10 ng/mL) of lipopolysaccharide (LPS), then the concentration of secreted TNF-α in the supernatants and the levels of TNF-α messenger RNA (mRNA) in BMDMs were examined. The results showed that there was no significant difference in the mRNA level, but that there was a significant decrease in TNF-α secretion from NRDC-deficient BMDMs at 3 hours after stimulation (figure 3B,C). Because impairment of TNF-α shedding leads to accumulation of membrane-bound TNF-α,35 we analysed the cell surface expression of TNF-α on BMDMs by flow cytometry and found that membrane-bound TNF-α was increased in BMDMs from NrdcdelM mice (figure 3D). ADAM17 is essential for TNF-α shedding, and we previously reported that NRDC enhances the sheddase activity of ADAM17 via a direct interaction. Consistent with this, there were no significant differences in ADAM17 mRNA levels between BMDMs from Nrdcflox/flox and NrdcdelM mice (figure 3E).
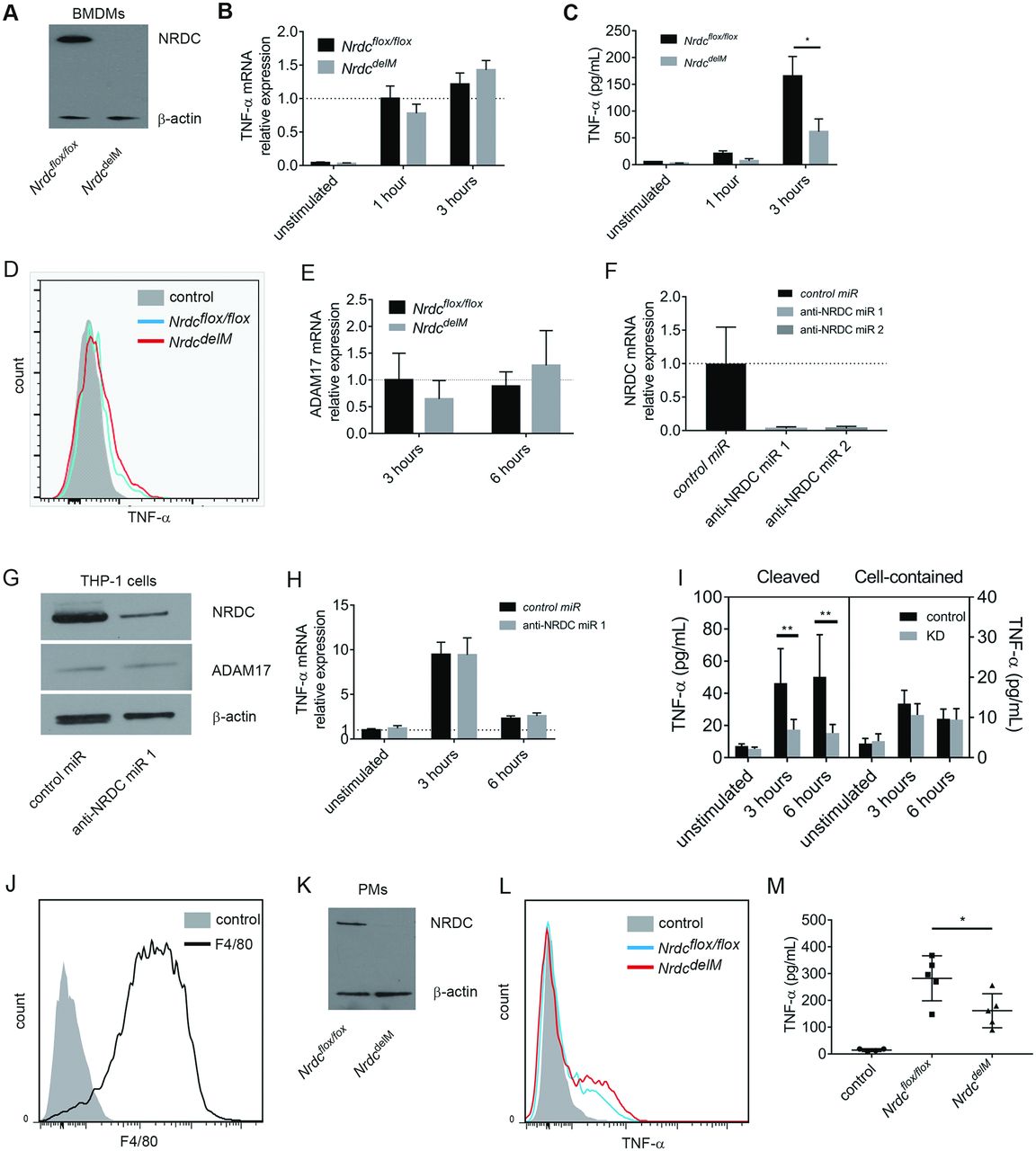
Secretion of TNF-α from macrophages and monocytic cells were reduced by the deletion of Nrdc. (A) Western blot analysis of NRDC in BMDM from Nrdcflox/flox (left) and NrdcdelM mice (right). β-Actin was used as a loading control. (B) TNF-α mRNA levels in unstimulated or LPS-stimulated BMDMs were analysed by reverse transcription and quantitative PCR (qRT-PCR). β-Actin mRNA levels were used as internal control. n=6 (mean ± SEM). (C) TNF-α secreted from BMDMs in conditioned medium was measured by ELISA. n=6 (mean ± SEM). (D) Membrane-bound TNF-α in BMDMs was stained 3 hours after LPS stimulation and analysed by flow cytometry. Filled histogram, unstimulated control; blue open histogram, Nrdcflox/flox ; red open histogram, NrdcdelM . Representative histograms out of four independent experiments are shown. (E) ADAM17 mRNA levels in unstimulated or LPS-stimulated BMDMs were analysed by qRT-PCR. β-Actin mRNA levels were used as internal control. n=6 (mean ± SEM). (F) NRDC mRNA levels in THP-1 cells, transfected with lentiviral vectors for negative control RNA interference (RNAi) (control microRNA (miR)) or RNAi targeting Nrdc (anti-NRDC miR), were analysed using qRT-PCR. β-Actin mRNA was used as an internal control. n=6 (mean±SEM). (G) NRDC and ADAM17 protein expressions in THP-1 cells transfected with lentiviral vectors for control miR (left) or anti-NRDC miR 1 (right) were analysed by Western blot. β-Actin was used as a loading control. (H) TNF-α mRNA levels in THP-1 cells, transfected with lentiviral vectors as described in (G), were analysed using qRT-PCR. n=8 (mean ± SEM). (I) Secreted TNF-α (conditioned medium) and cell-contained TNF-α (cell extracts) in THP-1 cells described above were measured by ELISA. n=8 (mean±SEM). (J) F4/80 expression in peritoneal macrophage (PMs). Filled histogram, isotype control; open histogram, F4/80. Representative histograms out of three independent experiments are shown. (K) NRDC expression in PMs, isolated from Nrdcflox/flox (left) and NrdcdelM mice (right) after the reverse passive Arthus reaction, was examined by Western blot. β-Actin was used as a loading control. (L) Membrane-bound TNF-α was stained and analysed by flow cytometry. Filled histogram, unstimulated control; blue open histogram, Nrdcflox/flox ; red open histogram, NrdcdelM . Representative histograms out of three independent experiments are shown. (M) Measurement of secreted TNF-α concentration in the peritoneal lavage of Nrdcflox/flox (n=5), NrdcdelM (n=5) and control (n=4) mice. *p<0.05; **p<0.01, Welch’s t-test.
We next assessed the effect of NRDC gene knockdown in a human monocytic cell line, THP-1. Efficient inhibition of NRDC mRNA and protein levels was confirmed in THP-1 cells infected with lentiviral vectors expressing RNAi targeting NRDC (figure 3F,G). LPS-induced TNF-α secretion in the supernatants was significantly decreased by gene knockdown of NRDC, whereas TNF-α mRNA levels and TNF-α protein concentrations in the cell lysates did not differ between control and NRDC-knocked down cells (figure 3H,I). Together, our results in mouse macrophages and human monocytic cells suggested that TNF-α activation via ectodomain shedding was inhibited by the deletion of NRDC.
To assess the in vivo role of NRDC on TNF-α secretion in a way more relevant to disease, we examined the effect of immunocomplexes (ICs) on TNF-α secretion. In the mechanism of AAIA, inflammatory cells are activated by ICs that are deposited in the joints. To mimic this response, we applied a reverse passive Arthus reaction peritonitis36 model to Nrdcflox/flox and NrdcdelM mice. In this model, ovalbumin (OVA) and anti-OVA antibody are administered separately, and then combine to make ICs and trigger complement pathways. We confirmed that cells collected from peritoneal lavage 3 hours after the administration were mostly F4/80 positive, and therefore were peritoneal macrophages (PMs) (figure 3J). We also confirmed that NRDC expression was markedly reduced in PMs from NrdcdelM mice (figure 3K), and analysis by flow cytometry demonstrated that membrane-bound TNF-α was increased in PMs from NrdcdelM mice (figure 3L). In contrast, secreted TNF-α in the peritoneal lavage was lower in NrdcdelM mice than in Nrdcflox/flox mice (figure 3M). These results indicated that ectodomain shedding of TNF-α was impaired in NRDC-deficient macrophages.
NRDC increased in synovial fluid of patients with RA
To assess the pathophysiological role of NRDC in patients, we collected synovial fluid from patients with RA or OA during joint surgery. Intriguingly, we found a remarkable and significant elevation of the level of NRDC and TNF-α in synovial fluid from RA patients compared with that from OA patients (table 1, figure 4A,B).
Patients’ characteristics
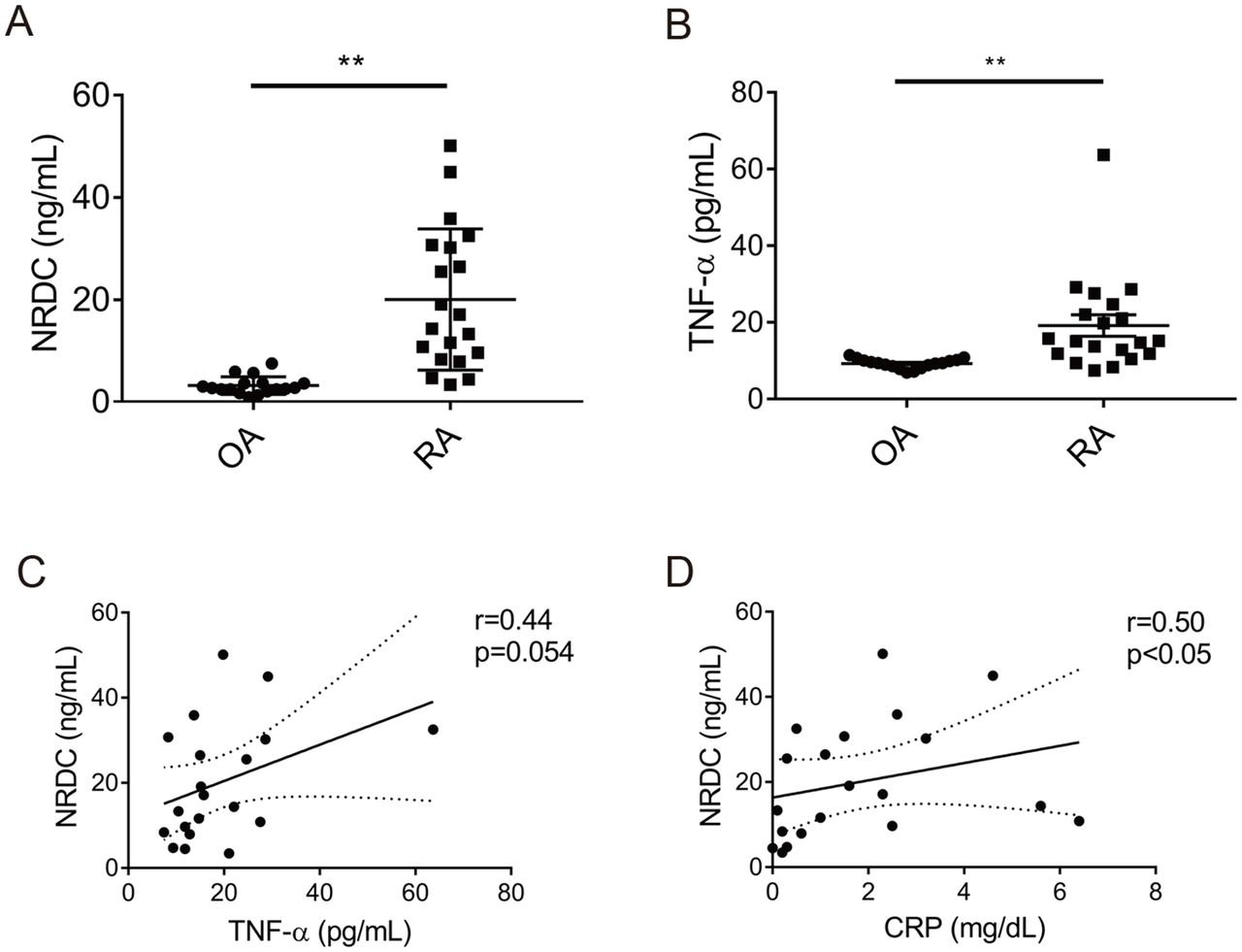
NRDC and TNF-α increase in synovial fluid from patients with RA. (A) NRDC concentration in synovial fluid from OA (n=17) and RA (n=20) patients. **p<0.01, Welch’s t-test. (B) TNF-α concentration in synovial fluid from OA and RA patients. **p<0.01, Welch’s t-test. (C) NRDC in synovial fluid has a tendency of positive correlation with TNF-α in RA patients. Solid line, approximate line; broken line, 95% CI. r=0.44, p=0.054, Spearman’s rank correlation test. (D) NRDC in synovial fluid is significantly correlated with CRP in RA patients. Solid line, approximate line; broken line, 95% CI. r=0.50, p<0.05, Spearman’s rank correlation test.
Gene silencing of Nrdc in the ankle joints ameliorated CAIA in WT mice
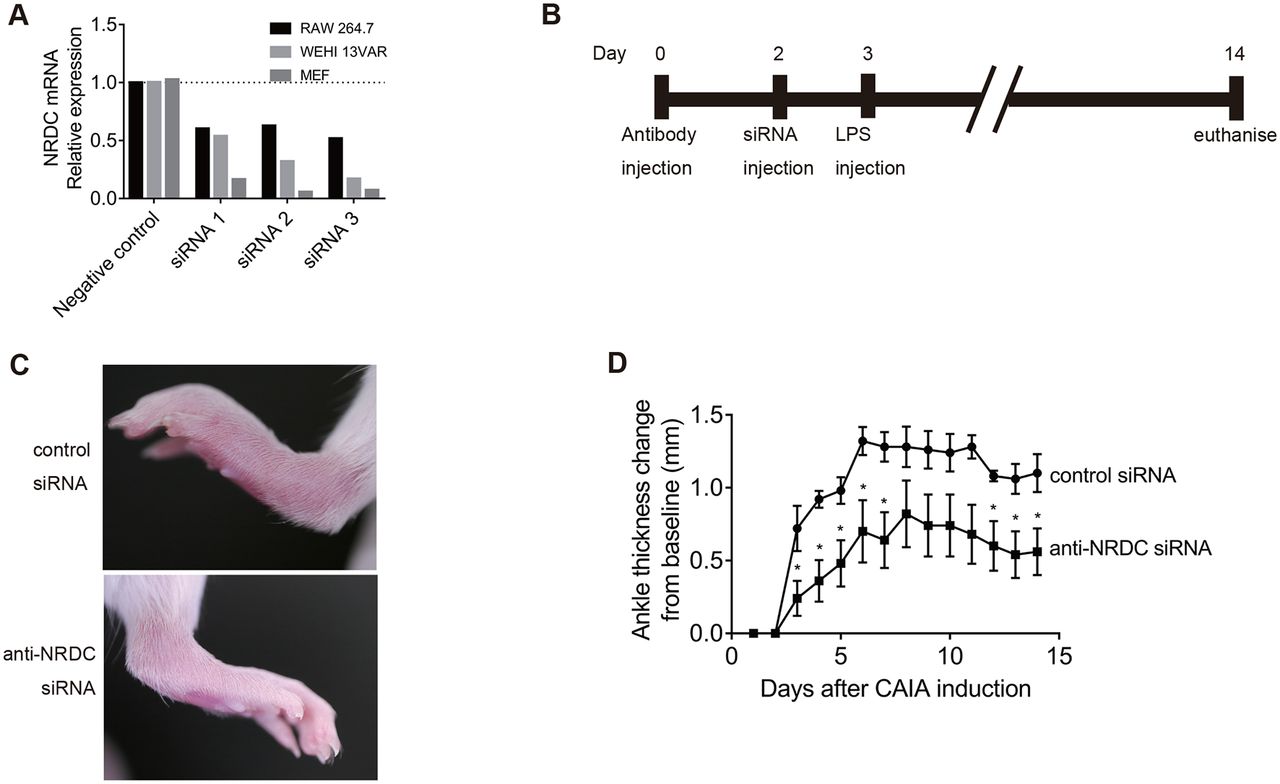
To confirm the pathological role in vivo of NRDC in autoimmune arthritis, we examined whether local gene silencing of Nrdc ameliorated CAIA. We first tested three siRNAs targeting Nrdc in three different cell lines (RAW264.7, WEHI 13 VAR and mouse embryonic fibroblasts) and selected the siRNA with the best silencing efficacy (siRNA3) (figure 5A). This anti-Nrdc siRNA was injected into the left ankle joint of mice 2 days after CAIA induction, while negative control siRNA was injected into the right ankle joint. The results showed that the severity of arthritis was clearly reduced in the left joint (anti-NRDC siRNA) compared with the right joint (control siRNA) (figure 5C). The measurement of ankle thickness revealed that anti-Nrdc siRNA significantly ameliorated CAIA from days 3 to 8 and days 12 to 14 (figure 5D).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Intra-articular Nrdc gene silencing with siRNA attenuates CAIA. (A) Screening of siRNAs against NRDC in RAW264.7, WEHI 13 VAR and mouse embryonic fibroblasts (MEF). (B) Schedule of the induction of CAIA and intra-articular injection of siRNA. (C) Photographs of the hind paws 4 days after the induction of CAIA. Anti-NRDC siRNA was injected in the left ankle, while control siRNA was injected in the right ankle joints of BALB/c WT mice. (D) Ankle thickness was measured daily after the induction of CAIA (n=5 per treatment). *p<0.05, Welch’s t-test.
Discussion
In this study, we demonstrate for the first time in vivo that NRDC plays important roles in autoimmune arthritis. First, Nrdc –/– mice were resistant to CAIA, and resistance to two models of AAIA, CAIA and K/BxN STA, was confirmed in NrdcdelM mice, indicating that NRDC in macrophages is crucial for the pathogenesis of autoimmune arthritis. In both CAIA and K/BxN STA models, inhibition of TNF-α has been shown to attenuate arthritis.4 18 Given that macrophages are a major source of TNF-α in the joints, reduced release of TNF-α from NRDC-deficient macrophages may strongly contribute to the attenuation of arthritis.6
TNF-α is synthesised as a type 2 membrane protein precursor, which is cleaved by ADAM17 at a juxtamembrane domain to be released from the cell surface. A comparison of the phenotypes of TNF-α-null mice and knockin mice expressing an uncleavable form of TNF-α (mice without soluble TNF-α) revealed that soluble TNF-α is dispensable for the organogenesis of most lymphoid tissues, but is indispensable for optimal development of inflammatory lesions.19 This finding indicated that TNF-α activation by ectodomain shedding plays important roles in inflammatory diseases. Indeed, myeloid cell-specific ADAM17-deficient mice were protected from K/BxN STA.34 Moreover, the deletion of iRHOM2, a critical regulator of protein maturation and of the cell surface localisation of ADAM17, also protected against K/BxN STA,34 further indicating that ADAM17-mediated TNF-α shedding is involved in AAIA. Our previous studies have demonstrated that NRDC enhances ectodomain shedding of TNF-α via activation of ADAM17.23 28 30 NRDC is one of the rare proteins that have been shown to bind directly to ADAM17 and enhance its enzymatic activity. Interestingly, the peptidase activity of NRDC is not required for the enhancement of ADAM17 activity.22–24 These previous findings prompted us to examine the role of NRDC in autoimmune arthritis, and we found that inhibition of NRDC had a protective effect against the disease. We have demonstrated here that macrophage-specific deletion of NRDC closely recapitulated the resistant phenotype of whole-body knockout mice in AAIA models. Using BMDMs, PMs and THP-1 cells, we showed that NRDC regulates ectodomain shedding of TNF-α in macrophages and monocytic cells. A deficiency of NRDC in all cell types resulted in a decrease in cleaved TNF-α, whereas TNF-α mRNA was not significantly affected. These results strongly suggest that NRDC in macrophages is involved in the pathogenesis of AAIA via the regulation of TNF-α shedding.
It was previously reported that the level of TNF-α is increased in RA synovial fluid.37 We demonstrate here that the level of NRDC and TNF-α in synovial fluid is remarkably elevated in RA patients compared with that in OA patients. NRDC levels in RA synovial fluid correlated with synovial fluid TNF-α and serum CRP, suggesting that a local increase of NRDC in the RA joint may enhance TNF-α secretion, resulting in aggravation of joint inflammation. These results suggest that NRDC plays important roles not only in mouse experimental models of RA but also in RA patients, and therefore that gene-targeted therapy with RNAi has potential in the treatment of RA.38 Intra-articular injection of anti-TNF-α siRNA was shown to inhibit collagen-induced arthritis in mice,39 and intravenous administration of double-stranded miR-451 reduced arthritis in SKG mice via downregulation of neutrophil chemotaxis.40 We show here that intra-articular injection of anti-Nrdc siRNA ameliorated CAIA. In this experiment, arthritis was induced more rapidly than without intra-articular injection, probably because of the inflammatory response against injected reagents or trauma associated with the injection. This significant attenuation of arthritis by anti-Nrdc siRNA provided an indication of a novel treatment for RA. Although biologic TNF-α blockers are widely used for the management of RA patients, complete inhibition of TNF-α might cause adverse effects, mainly infections, because a physiological level of TNF-α secretion is necessary for the normal inflammatory response. As we show here, gene deletion or silencing of NRDC in macrophages and monocytic cells did not affect their baseline level of constitutive TNF-α production and secretion. Thus, anti-NRDC therapy could have the potential to reduce only excessive TNF-α secretion. In a similar sense, ADAM17 could be an alternative target for controlling TNF-α secretion. However, ADAM17 cleaves not only TNF-α but also a wide range of membrane proteins including EGF receptor ligands and Notch,41 which may cause side effects of ADAM17 inhibitor treatment in the skin, intestine and liver.34 42 In terms of substrates, NRDC also enhances ectodomain shedding of various membrane proteins including HB-EGF, neuregulin1 and amyloid precursor protein.22 24 29 Other than TNF-α, several membrane proteins considered to be important for RA pathogenesis, such as TNF receptors and IL-6 receptors, are also functionally regulated by ectodomain shedding. NRDC-mediated regulation of the shedding of these molecules might contribute to the attenuation of arthritis by the deletion of NRDC.
Apart from its extracellular function as an enhancer of ectodomain shedding, a nuclear function of NRDC as a transcriptional coregulator has been recently clarified. The roles of NRDC in transcriptional regulation were illustrated by the interaction of NRDC with various nuclear proteins such as histone deacetylase 3, peroxisome proliferator-activated receptor gamma coactivator 1-α and islet1.43–45 Intriguingly, the nuclear function of NRDC is crucially involved in adaptive thermogenesis44 and insulin secretion in vivo.43 Although we cannot exclude a possible involvement of nuclear NRDC in AAIA, we demonstrate here that mRNA levels of TNF-α and ADAM17 in macrophages are not affected by the inhibition of NRDC, suggesting that it is extracellular NRDC that mainly regulates AAIA.
Another limitation of this study is that we have not clarified NRDC function in neutrophils, which have a role in the pathogenesis of AAIA.33 Because the Lyz2 gene is expressed not only in macrophages but also in neutrophils,46 NRDC in neutrophils should be deleted in NrdcdelM mice.
In summary, our results demonstrated for the first time that NRDC is involved in AAIA at least partially via enhancement of TNF-α shedding in macrophages. NRDC levels in synovial fluid are increased in RA patients and correlate with systemic inflammation, providing a new insight into the pathogenesis of RA. In addition, we showed that Nrdc gene silencing ameliorates arthritis in mouse experimental models of RA, suggesting that NRDC could be a new therapeutic target for RA.
Acknowledgments
We gratefully acknowledge the patients who consented to synovial fluid and blood collection for this study. Detection of human NRDC in synovial fluid was supported by Rina Yamaguchi, Yuto Kurokawa and Yoshiyuki Amano (Sanyo Chemical Industries). We thank Sayaka Saijo, Shintaro Matsuda, Hideyuki Shibuya, Hiromi Iwai, Ryoko Nakanishi and Yoshiko Nakajima (Kyoto University) for their excellent help. We are also grateful to Diane Mathis and Christophe Benoist (Harvard Medical School) for providing us with KRN mice.
References
Footnotes
Contributors TF, HI and EN designed the studies. TF and EN wrote the manuscript. TF, HY, MO, KN, Yus M, Yug M, AO and TT performed the experiments and analysed the data. NO harvested K/BxN mice serum. TF, HI, MF and MA were involved in the analysis of human subjects. TK and SM supervised the works. All authors read and approved the final manuscript.
Funding This work was supported by Grants-in-Aid for Scientific Research (KAKENHI) (26293068, 25462368, 15K19513, 15K19376, 15H01557, 16K15216, 16K108980004). It was also supported by the Takeda Science Foundation and the Uehara Memorial Foundation.
Competing interests None declared.
Patient consent Obtained.
Ethics approval The ethics committee of Kyoto University Graduate School of Medicine.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All available data can be obtained by contacting the corresponding authors.