Article Text

Abstract
Objective Verinurad (RDEA3170) is a high-affinity, selective URAT1 inhibitor in development for treating gout and asymptomatic hyperuricaemia. This study evaluated the pharmacodynamics, pharmacokinetics and safety of verinurad in combination with febuxostat in adults with gout.
Methods The phase IIa, open-label, multicentre study randomised 64 subjects into one of five cohorts to receive febuxostat (40 or 80 mg) alone or in combination with verinurad 2.5–20 mg. Serial plasma/serum and urine samples were assayed for verinurad and uric acid. Safety was assessed by adverse events, chemistry panels, ECGs and physical examinations.
Results Serum pharmacodynamic data demonstrated the maximum percent decrease in serum urate (sUA) from baseline (Emax) at 8–12 hours after dosing. Verinurad with febuxostat decreased sUA in a dose-dependent manner. Emax for verinurad with febuxostat 40 mg ranged from 52% to 77% vs 42% for febuxostat 40 mg alone; Emax for verinurad with febuxostat 80 mg was 62%–82% vs 55% for febuxostat 80 mg alone. Urinary uric acid excretion rate was reduced below baseline by febuxostat alone and was comparable to baseline for verinurad with febuxostat. Verinurad plasma exposure increased with dose and was comparable when combined with febuxostat. No drug-drug interactions were observed. Verinurad was well tolerated with no clinically meaningful changes in laboratory values.
Conclusion Verinurad administered with febuxostat produced dose-dependent decreases in sUA while maintaining urinary uric acid levels comparable to baseline. These dose combinations of verinurad and febuxostat were generally well tolerated. These data support continued investigation of oral verinurad in patients with gout.
Trial registration number NCT02246673
- febuxostat
- gout
- verinurad
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Febuxostat is a xanthine oxidase inhibitor (XOI) used to reduce serum urate (sUA) levels in patients with gout.
In patients not achieving target sUA levels on an XOI alone, guidelines recommend combining an XOI with a uricosuric agent.
What does this study add?
Verinurad is a potent uricosuric acid in development for chronic treatment of gout and asymptomatic hyperuricaemia.
Verinurad (doses 2.5–20 mg) combined with febuxostat (40 or 80 mg) produced dose-dependent decreases in sUA that were greater than with febuxostat alone, while maintaining urinary uric acid levels comparable to baseline.
How might this impact on clinical practice?
Verinurad in combination with an XOI may, if approved, represent an effective treatment option in patients with gout with an incomplete response to an XOI alone, providing greater sUA lowering than by escalating the dose of XOI monotherapy.
Introduction
Gout is a common, chronic inflammatory arthritis usually associated with excruciating pain,1 2 which is characterised by the deposition of monosodium urate crystals in the joints, tendons and other connective tissues, generally secondary to long-standing hyperuricaemia.3–5 Sustained reduction in the concentration of serum urate (sUA) below its saturation point leads to the dissolution of crystals with resultant alleviation of gout symptoms.6 7 Clinical trial experience shows that greater lowering of sUA is associated with improved outcomes in terms of tophus reduction and gout flare incidence.8–12
Urate-lowering treatments (ULTs) reduce sUA by different mechanisms.5 The xanthine oxidase inhibitors (XOIs), allopurinol and febuxostat, reduce the production of urate, while the uricosurics including probenecid, benzbromarone, sulfinpyrazone and lesinurad inhibit the reabsorption of urinary uric acid in the renal tubules to lower sUA levels. A substantial proportion of patients fail to achieve the target sUA (6 mg/dL, or 5 mg/dL for patients with severe gout symptoms) using an initial XOI as monotherapy.13 14 For these patients, treatment guidelines recommend switching to the alternative XOI or combining treatments with complementary mechanisms of action, such as an XOI with a uricosuric.7 15
Lesinurad was recently approved in combination with an XOI for the treatment of hyperuricaemia associated with gout in patients who fail to achieve target sUA on an XOI alone.16–18 The phase III clinical development programme of lesinurad included combination treatment with febuxostat 80 mg.17 Lesinurad at 200 and 400 mg doses in combination with febuxostat resulted in more patients achieving target sUA and experiencing reduction in overall tophus area compared with febuxostat alone.17 Neither of the lesinurad dose combinations significantly reduced gout flare rates when compared with febuxostat alone at 12 months, although there were numerical differences in favour of the 400 mg combination. However, the 400 mg lesinurad dose was associated with increased rates of renal adverse effects compared with lesinurad 200 mg plus febuxostat or febuxostat alone, leading to the decision to seek the US and European regulatory approval for only the 200 mg dose.
Verinurad is a uricosuric under clinical investigation that demonstrates high potency in inhibiting URAT1 in vitro19 and significant sUA lowering at doses as low as 2.5 mg in humans.20–22 The potency of verinurad, together with its pharmacokinetic (PK) properties, may enable it to be used at a low enough dose to have potentially less drug-drug interaction issues in patients with gout who receive concurrent treatments for comorbidities such as hypertension, cardiovascular disease, diabetes and obesity.
Verinurad was associated in monotherapy studies with an increase in urinary uric acid concentrations and low incidence of serum creatinine (sCr) elevation, as observed previously for other uricosurics.20 21 Febuxostat, by contrast, is known to reduce urinary uric acid excretion by inhibiting urate production.23 The combination of febuxostat with verinurad has the potential to lower sUA to a greater extent than by either treatment alone at the same or even higher monotherapy doses, while reducing the incidence of urinary uric acid elevation associated with verinurad monotherapy.24 25
The current study evaluated the pharmacodynamics (PD), PK and safety of multiple oral doses of verinurad in combination with oral febuxostat compared with febuxostat alone in adults with gout (NCT02246673). A notable feature of the study is the frequent urine samplings up to 24 hours postdose, to better characterise the urinary uric acid excretion profile of the verinurad and febuxostat combination.
Methods
Subjects
Male or female subjects with gout aged ≥18 and ≤75 years with body weight ≥50 kg, body mass index ≥18 to ≤45 kg/m2, sUA ≥8 mg/dL and estimated sCr clearance ≥60 mL/min (calculated by the Cockcroft-Gault formula using ideal body weight) were screened for enrolment. The subjects were diagnosed with gout according to the American Rheumatism Association Criteria for the Classification of Acute Arthritis of Primary Gout.26
The study was conducted in compliance with Good Clinical Practice and the Declaration of Helsinki. The subjects provided informed consent to participate in the study. The study was performed from October 2014 to July 2015.
Study design
Five cohorts of subjects were randomised into treatment sequences to receive oral verinurad at doses of 10 mg (cohort 1), 15 mg (cohort 2), 5 mg (cohort 3), 2.5 mg (cohort 4) and 10, 15 and 20 mg (cohort 5) in combination with febuxostat (40 or 80 mg) or febuxostat alone (40 or 80 mg) (figure 1). Randomisation used a centralised Interactive Web Response System that generated a unique randomisation number for each subject. Each treatment period was 7 days, which combined a period for washing out the previous treatment and for stabilising the new treatment. Treatments in cohorts 1 through 4 were conducted sequentially, while cohort 5 was conducted after review of the PD and PK results from cohort 2. Cohort 5 was designed such that subjects began at verinurad doses of 10 or 15 mg before receiving the highest investigated dose of 20 mg.

Study design. Febuxostat dose 40 or 80 mg; verinurad dose range 2.5–20 mg.
Subjects on prior ULT began gout flare prophylaxis with colchicine 0.6 mg at the beginning of ULT washout on approximately day −14, while subjects not on prior ULT started colchicine on approximately day −7. All study medications were administered once daily approximately 30 min after a standardised breakfast (approximately 650 kcal and 35% fat).
Blood and urine sampling
Serum samples for PD analyses were collected at screening, baseline (day –1) and weekly for 4 weeks within 30 min prior to dosing and at frequent preset intervals up to 24 hours postdose. Urine samples (total catch) for PD analyses were collected on day –1 and subsequently on day 1 (cohort 5 only) and weekly for 4 weeks at 1 hour up to 2 or 10 hours intervals.
Blood samples for PK analyses were collected weekly for 4 weeks within 30 min prior to dosing and at frequent intervals up to 24 hours postdose. Plasma was isolated from the blood by centrifugation. Urine samples for PK analyses were taken on days 14 and 21 (cohorts 1 through 4) and days 7, 14, 21 and 28 (as applicable to sequence assignment for cohort 5) at frequent intervals up to 24 hours postdose.
On urine collection days, subjects were instructed to drink 240 mL water immediately on waking in the morning (within 2 hours predose) in order to maintain urine output. Study medication was administered with another 240 mL water. After dosing, subjects were instructed to drink ~160 mL water/hour for the next 12 hours with an additional 240 mL at 22 hours.
Analytical methods
Verinurad concentrations in plasma and urine and febuxostat concentrations in plasma were measured by Ardea Biosciences (San Diego, California, USA). Plasma samples were extracted by protein precipitation and analytes quantified by high-performance liquid chromatography-mass spectrometry/mass spectrometry (LC/MS/MS).22 27 Urine samples were prepared by dilution with water and quantified by high-performance LC/MS/MS. Analyses of xanthine and hypoxanthine in urine samples were performed by inVentiv Health Clinique (Québec, Canada) by LC/MS/MS. Performance of the assays during study conduct was measured using quality control samples (QCs) prepared in a blank matrix containing analytes of interest. Verinurad human plasma QCs showed accuracy (measured by %relative error (%RE)) between –2.0% and 0.3%, while the precision of the QCs (measured by %coefficient of variation (%CV)) was ≤5.2%. Febuxostat plasma QCs demonstrated %RE values of –2.4 to –1.0%, while the %CV was ≤8.1%. Verinurad human urine QCs showed %RE values of –1.8% to 4.0% and ≤7.1%.
End points and determinations
All subjects who received at least one dose of verinurad or febuxostat and had evaluable PD or PK data were included in the PD and PK populations, respectively. All subjects who received at least one dose of verinurad or febuxostat made up the safety population.
The primary study objective was to assess the multiple-dose PD of verinurad in combination with febuxostat compared with febuxostat alone. PD parameters included maximum (Emax (%)) and mean time-matched percent change in sUA from baseline (day –1), lowest mean sUA (defined as the lowest absolute sUA level achieved by each treatment), rate of urinary uric acid excretion (ReUR), amount of urinary uric acid recovered (AeUR), renal clearance of uric acid (CLUR) and fractional excretion of uric acid (FEUA) for each time point for each 24-hour collection period. Urine samples were analysed for xanthine and hypoxanthine as a measure of the functional activity of febuxostat 40 mg.
For PD analyses, a mixed-effects model was used on sUA and urinary uric acid parameters, with treatment and period as fixed effects, subject as a random effect and baseline value as a covariate for each cohort. The least squares means for each treatment group and between-treatment differences, along with 95% CI and P values, were estimated. All statistical testing was conducted at an α level of 0.05. No adjustments were made for multiple comparisons. For summary statistics, data were pooled across cohorts according to treatment. PD data were collected by Covance Central Laboratory Services (Indianapolis, Indiana, USA) and PD analyses were performed using SAS V.9.3 or later (SAS Institute, Cary, North Carolina, USA).
PK assessments investigated the dose proportionality of verinurad 2.5–20 mg in the presence of febuxostat 40 or 80 mg. PK parameters for verinurad and febuxostat included maximum observed plasma concentration (Cmax), time of maximum observed plasma concentration (Tmax), area under the plasma concentration-time curve from time 0 to 24 hours postdose (AUC0-24), plasma terminal half-life (t½), amount excreted unchanged in urine (Ae0-24), fraction excreted in urine as unchanged drug or metabolite and renal clearance from time 0 to 24 hours (CLR,0-24).
For PK analyses, a mixed-effects model was used to assess the impact of febuxostat or verinurad dose on verinurad and febuxostat PK, respectively, with treatment as a fixed effect and subject as a random effect. PK analyses were conducted by Ardea Biosciences, using WinNonlin Professional V.6.3 (Pharsight, Mountain View, California, USA).
Subjects were monitored for safety throughout the study and at follow-up on day 42 (±2). Safety assessments included adverse events (AEs) coded according to the Medical Dictionary for Regulatory Activities (MedDRA, V.17.0), clinical laboratory evaluations, vital signs, ECGs and physical examinations. AEs were defined as serious if they resulted in death, were life-threatening, required hospitalisation or prolongation of existing hospitalisation or caused persistent or significant disability/incapacity. Any renal serious AEs were to be reviewed by a Renal Events Adjudication Committee appointed by the study sponsor during conduct of the verinurad clinical studies. Any haematology, chemistry or urinalysis abnormalities considered clinically relevant were to be assigned a severity rating by the investigator, based on Rheumatology Common Toxicity Criteria V.2.0 2007.28 Safety data are summarised by descriptive statistics using SAS V.9.3 or later (SAS Institute).
Results
Subjects
A total of 64 subjects were randomised to treatment sequences in one of the five cohorts (12–14 subjects per cohort), of whom 60 subjects completed the study. Four randomised subjects were withdrawn from the study because of high triglyceride levels at screening (n=1), non-compliance (n=1), loss to follow-up (n=1) and withdrawal of consent (n=1).
The demographic and baseline characteristics of the subjects are summarised in table 1. All subjects were males and the majority (67.2%) were white. Mean age (range 45–50 years) and body mass index (range 30.2–34.2 kg/m2) were similar across cohorts. The overall mean (SD) sUA at day –2 (baseline) was 9.3 (1.45) mg/dL.
Demographic characteristics and serum urate levels (safety population)
Pharmacodynamics
Serum urate
The time course of mean percent change in sUA from baseline (time-matched) is shown in figure 2. Maximum percent decreases in sUA from baseline (Emax) were observed between 8 and 12 hours after dosing for each treatment. Verinurad (2.5–20 mg) combined with febuxostat 40 mg and verinurad (2.5–15 mg) combined with febuxostat 80 mg decreased Emax in a dose-dependent manner (see online supplementary figure 1). Emax for varying doses of verinurad with febuxostat 40 mg ranged from 52% (verinurad 2.5 mg) to 77% (verinurad 20 mg) compared with 42% for febuxostat 40 mg alone, and Emax for verinurad with febuxostat 80 mg ranged from 62% (verinurad 2.5 mg) to 82% (verinurad 15 mg) compared with 55% for febuxostat 80 mg alone (P<0.05, all comparisons). Emax was greater for combinations of verinurad ≥5 mg with febuxostat 40 mg than for febuxostat 80 mg alone (see online supplementary figure 1).
Supplementary file 1

Mean (SE) percent change from baseline in sUA (µmol/L) following oral doses of febuxostat 40 mg (top) or 80 mg (bottom) in combination with varying doses of verinurad vs febuxostat 40 or 80 mg alone (overall treatment pooled across cohorts) (PD population. Patient n in parentheses: top (febuxostat 40 mg): febuxostat alone (60), verinurad 2.5 mg (12), verinurad 5 mg (12), verinurad 10 mg (24), verinurad 15 mg (23), verinurad 20 mg (11); bottom (febuxostat 80 mg): febuxostat alone (48), verinurad 2.5 mg (12), verinurad 5 mg (12), verinurad 10 mg (11), verinurad 15 mg (12)). PD, pharmacodynamics.
Verinurad in varying doses combined with febuxostat 40 or 80 mg consistently decreased the lowest mean sUA in a dose-dependent manner, consistent with Emax analyses (see online supplementary figure 2, pooled cohort; online supplementary table 1, data by cohort 1–4).
Urinary uric acid
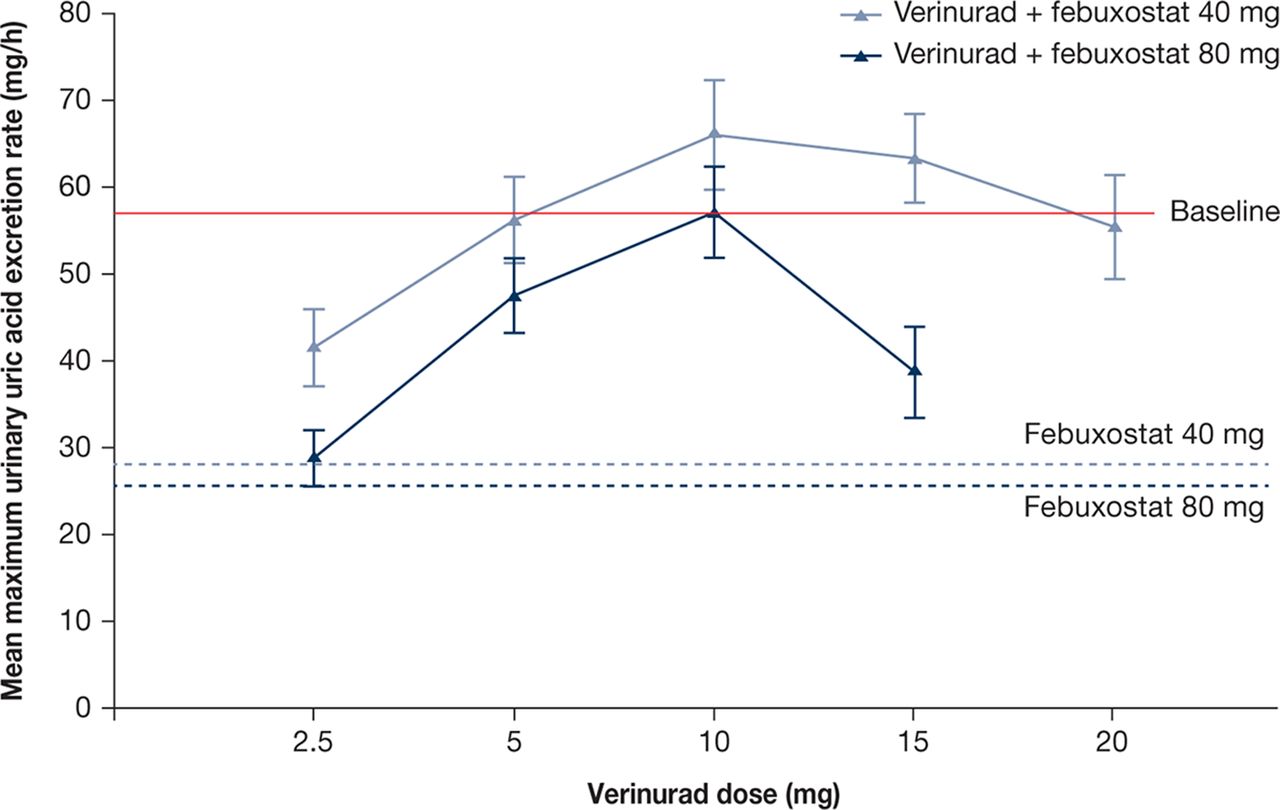
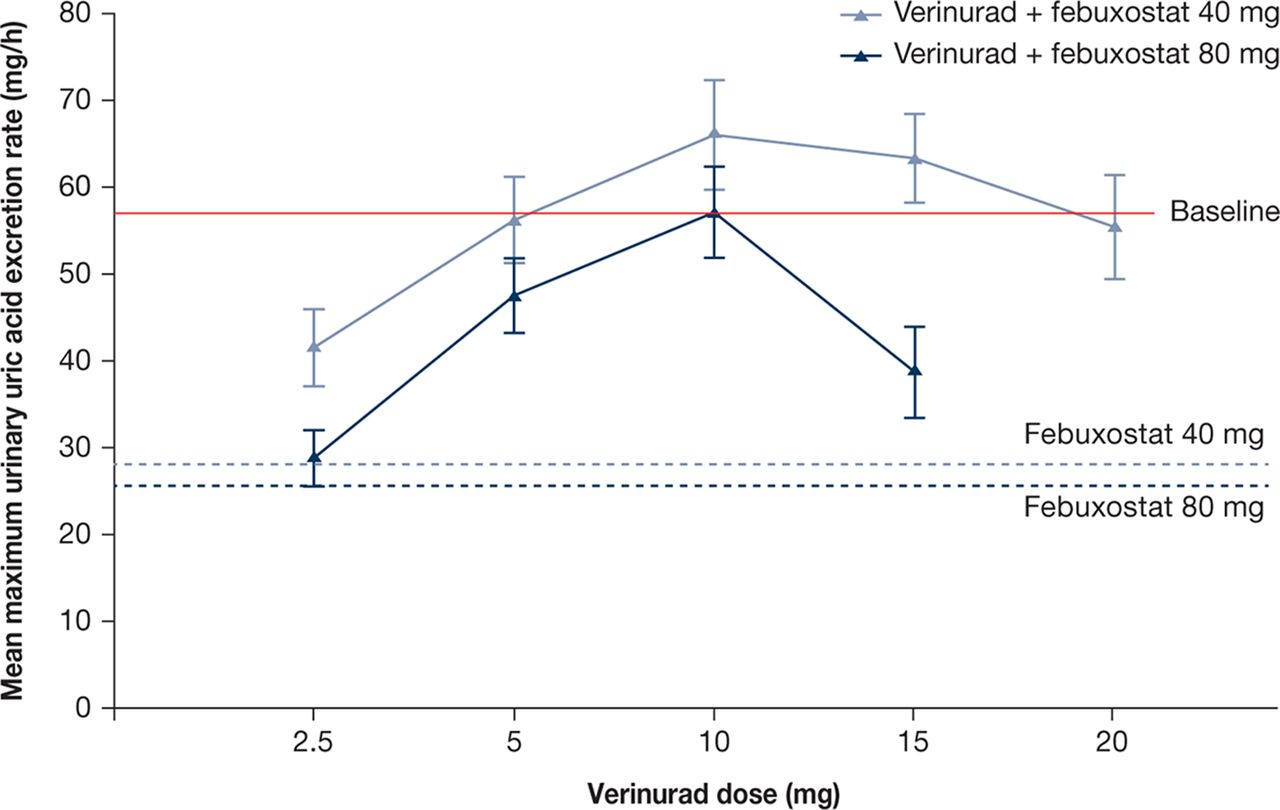
Mean maximum ReUR was reduced by febuxostat 40 and 80 mg compared with baseline (figure 3). Combinations of verinurad with febuxostat 40 or 80 mg resulted in a mean maximum Reur that was either lower than baseline (verinurad 2.5 and 5 mg) or comparable to baseline (verinurad 10, 15 and 20 mg).
{kind=link}
{kind=link}
{kind=link}
Mean (SE) maximum urinary uric acid excretion rate (mg/hour) following verinurad doses in combination with febuxostat 40 or 80 mg vs febuxostat 40 or 80 mg alone (pharmacodynamics population, pooled across cohorts. Patient n: see figure 2).
Mean AeUR over 0–12 or 12–24 hours for verinurad combined with febuxostat 40 or 80 mg was lower than the time-matched baseline and was comparable to febuxostat alone. Mean FEUA and CLUR were increased dose dependently by verinurad combined with febuxostat relative to baseline. Febuxostat 40 or 80 mg administered alone did not influence FEUA (online supplementary table 2) or CLUR (data not shown).
Urinary xanthine and hypoxanthine
Mean excretion of xanthine and hypoxanthine increased by 1965% and 527%, respectively, following febuxostat 40 mg alone, with comparable changes following febuxostat 40 mg combined with verinurad 10, 15 or 20 mg (data not shown).
Pharmacokinetics
Mean plasma concentration profiles of verinurad at doses of 2.5, 5, 10, 15 and 20 mg in combination with febuxostat 40 or 80 mg are shown in online supplementary figure 3 A,B. The median Tmax of verinurad ranged from 2.50 to 4.00 hours postdose, with a mean t1/2 from 8.26 to 13.2 hours (table 2). Increases in verinurad Cmax and AUC were dose dependent. Exposures of verinurad were generally similar when administered with febuxostat at either 40 or 80 mg doses.
Plasma PK of verinurad in the presence of febuxostat 40 or 80 mg (geometric mean (95% CIs)) (PK population)
Mean plasma concentration profiles of febuxostat at doses of 40 or 80 mg in combination with varying doses of verinurad are shown in online supplementary figure 3C,D. Increases in febuxostat Cmax and AUC0-24 were dose dependent. Comparisons of febuxostat exposures when administered alone or in combination with verinurad indicated equivalence (table 3).
Plasma PK of febuxostat in the presence of verinurad vs febuxostat alone (geometric mean (95% CIs)) (PK population)
The fraction of verinurad excreted in urine as unchanged drug ranged from 0.6% to 1.0%. Verinurad Ae0-24 increased with verinurad dose, from 15.5 µg (verinurad 2.5 mg with febuxostat 80 mg) to 143 µg (verinurad 20 mg with febuxostat 40 mg) and CLR,0-24 ranged from 8 to 12 mL/min with no evident relationship to verinurad dose. These parameters were not influenced by febuxostat dose.
Safety
Twenty subjects reported 30 treatment-emergent AEs (TEAEs) during treatment or follow-up (see online supplementary table 3). All TEAEs were mild or moderate in severity, with one exception (a severe case of hypertriglyceridaemia in the febuxostat 80 mg-alone group). There were no serious AEs, withdrawals due to AEs or renal-related events. The most frequent TEAE was pain in the extremity, reported in three subjects receiving verinurad and febuxostat combination treatment (see online supplementary table 3).
No clinically meaningful changes in laboratory values or vital signs were noted. There were no cases of sCr elevation ≥1.5× baseline.
Discussion
Verinurad is a potent, selective inhibitor of the URAT1 transporter that is under clinical development for the chronic treatment of gout and asymptomatic hyperuricaemia. This phase IIa, randomised study of 64 subjects with gout and sUA ≥8 mg·/dL showed that verinurad (2.5–20 mg) in combination with febuxostat (40 or 80 mg) reduced sUA in a dose-dependent manner. Maximum sUA reductions from baseline were ≥70% with verinurad doses of 10, 15 and 20 mg in combination with febuxostat 40 or 80 mg. Notably, at the higher doses investigated, the sUA lowering achieved (approximately 120 µmol/L) appeared comparable to the efficacy seen with injectable uricase therapies.29
Verinurad at doses ≥5 mg combined with febuxostat 40 mg also produced greater sUA lowering than febuxostat 80 mg alone, indicative of a greater PD effect for the combination than the higher dose of febuxostat monotherapy. Phase Ib studies of lesinurad (another URAT1 inhibitor) added to an XOI in patients with gout produced similar results by demonstrating that combination therapy produced greater sUA lowering than an increase in XOI dose,30 while studies of benzbromarone combined with allopurinol or febuxostat have demonstrated greater efficacy for combination treatment versus monotherapy.9 31–33
To fully characterise the rate of the urinary uric acid excretion post-treatment with verinurad, urine samples were collected as frequently as hourly. Such data are useful in understanding the risk for crystallisation of uric acid in urine and related AEs. The maximum rates of uric acid excretion in the combination groups were comparable to baseline levels, while uric acid excretion over 12–24 hours postdose was lower than baseline, which may be attributed to the febuxostat-mediated decrease in urate production.23 These observations suggest that the risk of uric acid crystallisation, manifesting as acute urate nephropathy and/or increased risk for nephrolithiasis, can be reduced or eliminated by co-administering febuxostat with verinurad.
Mean excretion of xanthine and hypoxanthine were increased by febuxostat, an observation consistent with its mechanism of action and in agreement with previous observations.34 This effect of febuxostat on xanthine and hypoxanthine excretion was largely unaffected by combination with verinurad. Plasma exposures of verinurad increased dose proportionally and were similar on co-administration of febuxostat 40 or 80 mg. These data together indicate an absence of drug–drug interaction between verinurad and febuxostat.
These dose combinations of verinurad and febuxostat were generally well tolerated, with no serious AEs or renal-related events and no clinically meaningful changes in laboratory evaluations during treatment.
A limitation of the study is the short study duration. Additional safety data will also be required in future studies. Strengths of this study include the investigation of subjects with gout and the frequent blood and urine samplings that enhanced the reliability of the PD and PK analyses. In particular, the frequent urine sampling allowed detailed assessment of the urinary uric acid excretion profile over time, which may be used to identify dose combinations of verinurad and febuxostat that mitigate the risk of urinary overexcretion of uric acid.
In conclusion, oral verinurad at the doses studied (2.5–20 mg) in combination with febuxostat (40 or 80 mg) produced superior sUA lowering than febuxostat alone in subjects with gout. Achieving low sUA levels is essential to reduce both frank tophi and other uric acid crystal deposits. All dose combinations of verinurad and febuxostat studied were generally safe and well tolerated. These data support continued investigation of oral verinurad in a dual-mechanism approach to lowering sUA.
Acknowledgments
Editorial support for this manuscript was provided by Bill Wolvey of PAREXEL, which was funded by AstraZeneca.
References
Footnotes
Contributors RF, PW, JH, SV, SL, XY, LH, CL, JNM, MG, MH-I: acquisition, analysis and interpretation of data for the work; drafting the work or revising it critically for important intellectual content and final approval of the version to be published. JH, SL, CL, JNM, MG: conception and design of the work.
Funding Funding for this study was provided by Ardea Biosciences/AstraZeneca.
Competing interests RF received a clinical study grant from Ardea Biosciences. PW is a full-time employee of Anaheim Clinical Trials. JH, SV, SL, XY, LH, CL and JNM are/were full-time employees of Ardea Biosciences, a member of the AstraZeneca Group at the time of this study. MG is a full-time employee of AstraZeneca.
Patient consent Obtained.
Ethics approval The protocol, protocol amendments and consent forms were approved by an Independent Ethics Committee/Institutional Review Board (Schulman Associates IRB, Cincinnati, Ohio, USA).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement De-identified patient data from this study are made available for scientific research on a case-by-case basis through AstraZeneca’s Data Request Portal: https://astrazenecagroup-dt.pharmacm.com//DT/Home/Index/