Article Text

Abstract
Background Baricitinib was efficacious in a 24-week phase III study in patients with rheumatoid arthritis (RA) and an inadequate response to conventional synthetic disease-modifying anti rheumatic drugs (DMARDs) (csDMARDs) (RA-BUILD).
Objectives To evaluate radiographic progression of structural joint damage in RA-BUILD patients over 48 weeks of baricitinib treatment in the long-term extension study, RA-BEYOND.
Methods In RA-BUILD, patients were randomised to placebo, baricitinib 2 mg or 4 mg once daily, with rescue possible from week 16. Patients completing RA-BUILD and entering RA-BEYOND continued to receive the baricitinib dose received at the end of RA-BUILD. Patients receiving placebo were switched to baricitinib 4 mg in RA-BEYOND. Joint damage was measured using the van der Heijde modified total Sharp score. To account for missing scores and scores obtained after rescue, switch or discontinuation of study drug, data were analysed using (1) linear extrapolation (LE) and (2) observed/last observation carried forward (LOCF). The observed/LOCF method used all available observed data, including after rescue or switch, with patients analysed according to original treatment assignment.
Results Using LE, radiographic progression at 24 and 48 weeks was statistically significantly lower for both baricitinib 2 or 4 mg compared with placebo. Only baricitinib 4 mg demonstrated statistically significant inhibition of progressive radiographic joint damage compared with patients initially randomised to placebo using observed/LOCF at week 48.
Conclusions Once daily oral baricitinib inhibited radiographic progression of structural joint damage in patients with an inadequate response or intolerance to csDMARDs over 48 weeks. The most robust benefit was seen for the 4 mg dose.
- rheumatoid arthritis
- radiographic progression
- RA-BUILD
- baricitinib
- csDMARD
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is already known about this subject?
In the phase III study RA-BUILD (Dougados et al Ann Rheum Dis 2017;76:88–95), baricitinib inhibited progression of radiographic joint damage over 24 weeks in patients with active rheumatoid arthritis who had an inadequate response to conventional synthetic disease-modifying antirheumatic drugs (DMARDs).
What does this study add?
This study demonstrates that treatment with baricitinib 2 and 4 mg maintains for up to 1 year the inhibition of structural joint damage originally seen at 24 weeks.
How might this impact on clinical practice?
These data show the sustained disease-modifying capacity of baricitinib.
The most robust benefit across measures was seen for the 4 mg dose.
Introduction
Rheumatoid arthritis (RA) is, if untreated, associated with progressive joint destruction, significantly compromised quality of life and reduced survival.1 2 The progressive joint damage caused by RA is associated with long-term functional disability3; therefore, a treatment goal for RA is to inhibit structural joint damage through the use of disease-modifying antirheumatic drugs (DMARDs). For a drug to be defined as a DMARD, it has to demonstrate that it is capable of inhibiting the rate of radiographic progression of structural joint damage.4
Baricitinib is an oral, selective, inhibitor of JAK1 and JAK2 and has demonstrated clinical benefit, including reducing the rate of structural joint damage, in several studies in patients with RA.5–8 In the 24-week phase III RA-BUILD study (NCT01721057), baricitinib significantly reduced radiographic progression of joint damage in patients with active RA who had an inadequate response to or were intolerant of previous conventional synthetic DMARDs (csDMARDs).8 On completion of the 24-week RA-BUILD study, patients could enrol in the long-term extension study, RA-BEYOND (NCT01885078). This report describes the effect of treatment with baricitinib 2 or 4 mg over 48 weeks on radiographic progression of structural joint damage in patients who completed RA-BUILD and enrolled in RA-BEYOND.
Methods
Patients
Patients who entered the RA-BUILD study were ≥18 years old with active RA (≥6/68 tender and ≥6/66 swollen joints; serum C reactive protein ≥3.6 mg/L (upper limit of normal 3.0 mg/L)), had an insufficient response (despite prior therapy) or intolerance to ≥1 csDMARDs and were biologic DMARD (bDMARD)-naïve. In the 24-week RA-BUILD study, patients were randomised to placebo, baricitinib 2 mg or baricitinib 4 mg, stratified by region and presence of joint erosions (yes/no) on centrally read radiographs obtained at screening. Rescue treatment (baricitinib 4 mg) was assigned at week 16 for patients whose tender and swollen joint counts improved from baseline by <20% at both weeks 14 and 16. For more details on the patient population that participated in the RA-BUILD study, please see Dougados et al’s study.8 Patients who completed the RA-BUILD study were eligible to enter the long-term extension study, RA-BEYOND. Rescue in RA-BUILD did not preclude enrolment in RA-BEYOND. Patients were not eligible for participation in RA-BEYOND if they demonstrated significant uncontrolled laboratory abnormalities, had a known hypersensitivity to baricitinib, or if they permanently discontinued baricitinib during the RA-BUILD study. Radiographs from RA-BUILD baseline, and weeks 24 and 48 in RA-BEYOND were scored in the reading campaign used to generate the data for these analyses.
Study protocol and oversight
RA-BEYOND is a phase III, multicentre, extension study evaluating the long-term efficacy and safety of baricitinib (2 mg once daily (QD) and 4 mg QD) in patients with RA.
Patients who completed RA-BUILD and entered RA-BEYOND continued to receive the dose of baricitinib they were receiving at the end of RA-BUILD (baricitinib 2 or 4 mg). Patients receiving placebo at the end of RA-BUILD were switched to baricitinib 4 mg on entry into RA-BEYOND. Although patients and investigators were aware that all patients would be receiving baricitinib in the extension study, those who had not been rescued remained blinded to randomised treatment assignment from RA-BUILD. In RA-BEYOND, patients could continue to receive background open-label csDMARDs, non-steroidal anti-inflammatory drugs, analgesics and/or corticosteroids (≤10 mg of prednisone or equivalent per day) being received at the completion of RA-BUILD. Patients with a Clinical Disease Activity Index (CDAI) score >10 at or after 3 months following enrolment into BEYOND could be rescued to baricitinib 4 mg. After rescue, all patients would receive 4 mg and remain blinded to their previous treatment assignment. Investigators were free to prescribe new csDMARDs and add or increase dosage of steroids, NSAIDS and/or analgesics. Baricitinib 4 mg was open label after rescue. Use of bDMARDs was prohibited in RA-BEYOND.
The RA-BEYOND study was designed by the sponsor, Eli Lilly and Company, an academic advisory board including non-Lilly authors of this manuscript, and Incyte Corporation. It was conducted in accordance with ethical principles of the Declaration of Helsinki and Good Clinical Practice guidelines and approved by the institutional review board or ethics committee for each centre. All patients provided written informed consent before the first study procedure. The study commenced in June 2013 and was ongoing at the time the present manuscript was prepared. Lilly or its representatives provided data, laboratory and site-monitoring services. All authors participated in data analysis and interpretation, reviewed drafts and final manuscript, and provided critical comments. The authors vouch for the veracity and completeness of the data and data analyses.
Efficacy
Radiographic joint damage was measured using the van der Heijde modified Total Sharp Score (mTSS).9 10 This methodology quantifies the extent of bone erosions and joint space narrowing for 44 and 42 joints, respectively, within the hands and feet with higher scores representing greater damage. Radiographs were scored by two primary readers separately and independently, blinded to chronological order, patient identity and treatment group. The mean score obtained between the two readers was used in the analyses.5 6 Radiographic progression was determined by the change from baseline in mTSS, bone erosion and joint space narrowing. Progression versus non-progression was defined based on change from baseline in mTSS using thresholds of 0, 0.5 or the smallest detectable change (SDC). The initial radiographs taken at RA-BUILD baseline served as the baseline radiographs for comparison throughout RA-BEYOND. The time points presented in this manuscript were all relative to the RA-BUILD baseline. Radiographs were obtained at baseline, weeks 24 and 48. The 48-week radiographs were obtained during the RA-BEYOND study. If the patient was rescued or had an early termination visit, radiographs were obtained only if the most recent radiographs were >12 weeks earlier. All these radiographs were analysed in a single reading campaign.
Safety
Clinical laboratory tests, vital signs and other safety assessments were performed at scheduled visits. The occurrence and severity of all adverse events (AEs) were recorded.
Statistical analyses
Analysis of structural progression was conducted on the modified intent-to-treat population using patients with available baseline (from RA-BUILD study) and at least one radiographic assessment at any time after baseline. Analysis of covariance (ANCOVA) was used to analyse radiographic progression, and a logistic regression model was used to analyse percentage of patients with no progression of structural joint damage, with baseline value (for ANCOVA model only), treatment, region and centrally confirmed presence of baseline joint erosions in the model. The Fisher exact test was used for categorical safety data or when sample size requirements for the aforementioned logistic regression model were not met. Analyses were assessed with a significance level of 0.05 (two-sided). The SDC in mTSS was computed from the variability in week 0 to 24 or to week 48 (1 year) changes in scores assigned by the two blinded readers.11
Patients who were rescued or discontinued in either RA-BUILD or RA-BEYOND, or did not enter RA-BEYOND, were defined thereafter as non-responders (non-responder imputation) for categorical clinical efficacy outcomes. To account for missing mTSS and data obtained after rescue or treatment switch, data were analysed using (1) linear extrapolation (LE) and (2) observed/last observation carried forward (LOCF). The LE method used radiographic data at baseline and the last radiograph taken before discontinuation, rescue or switch to impute missing data. The observed/LOCF method used all available observed data, including after rescue or switch, with patients analysed according to original treatment assignment. The result obtained from the last obtained radiograph after baseline was used for subsequent timepoints. Safety observations were analysed by assigned treatment until the time of rescue or switch or completion of the treatment period. P values were not adjusted for multiple comparison.
Results
Patients
In RA-BUILD, 684 patients were randomised, and 611 patients completed the study; 583 patients entered RA-BEYOND (85% of the randomised and 95% of the completed patients). Baseline demographics and clinical characteristics were similar among treatment groups.8 See figure 1 for the number of patients who were rescued and a brief description of rescue criteria. The distribution of patients included in the LE and observed/LOCF analyses can be seen in figure 2. Patients who remained on baricitinib 2 mg in RA-BUILD continued on 2 mg in RA-BEYOND. Patients who were rescued from baricitinib 2 to 4 mg, or who were originally randomised to placebo or baricitinib 4 mg, received baricitinib 4 mg in RA-BEYOND.

Patient disposition through 48 weeks in RA-BEYOND. Patients who were rescued or discontinued from the originator study, RA-BUILD, or who completed RA-BUILD, entered RA-BEYOND and were rescued or discontinued from RA-BEYOND on or before 48 weeks, were defined as non-responders or had their last observations before rescue or discontinuation used for analyses of subsequent time points for efficacy endpoints. *Patients on placebo at the end of the RA-BUILD study were switched to baricitinib 4 mg on entry to RA-BEYOND. LTE, long-term extension; QD, once daily; RA, rheumatoid arthritis.
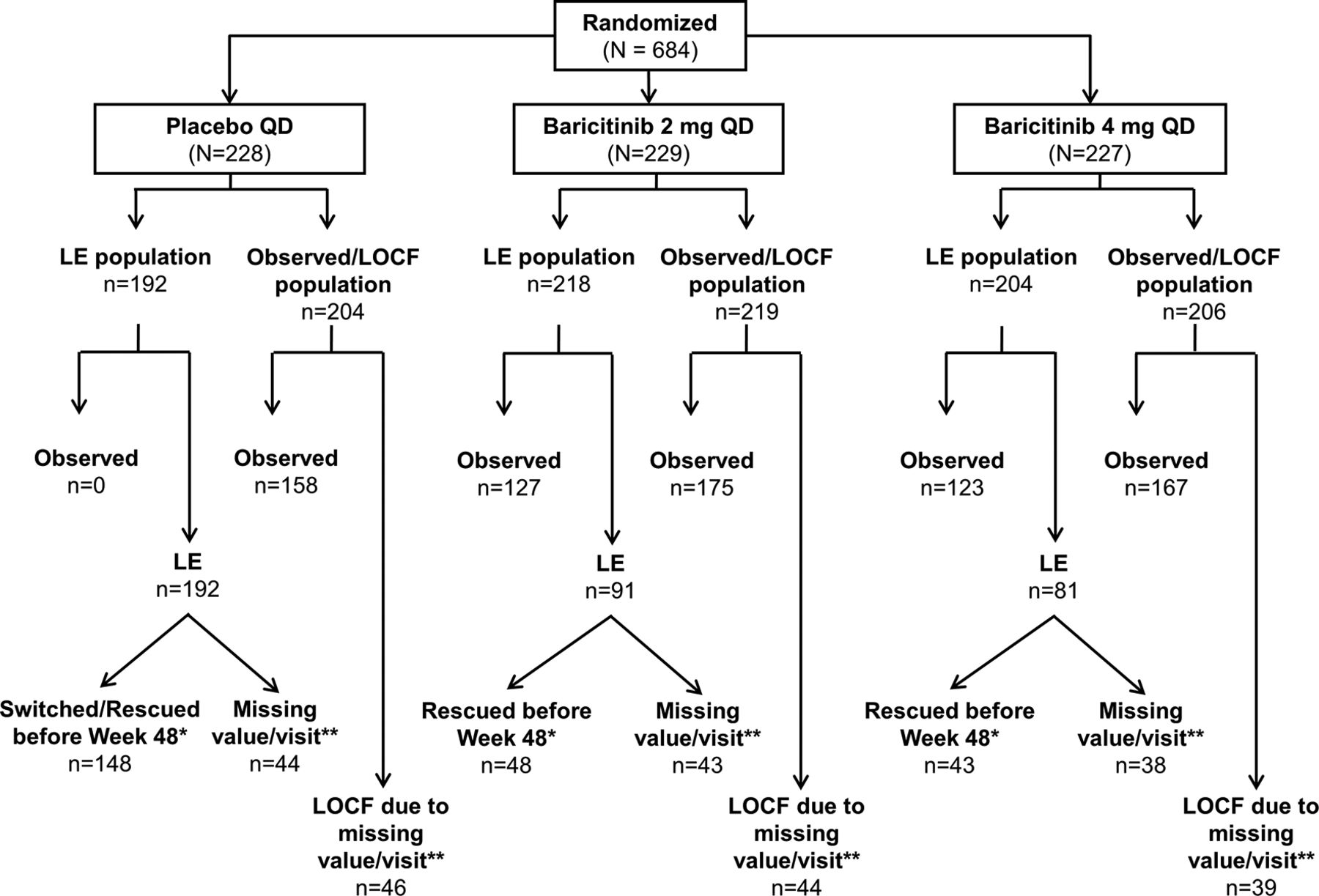
Patients included in linear extrapolation and observed/last observation carried forward analyses at week 48. Patients who were rescued or discontinued in RA-BEYOND were defined as non-responders. Missing scores and scores obtained after rescue or discontinuation were analysed using both LE and observed/LOCF. *LE imputations were performed for patients who had non-missing values at Week 48 but had been rescued before week 48 or switched at week 24 (placebo responders). If a patient fit into more than one reason, the reason with the earliest date was used. **This value represents all patients with a missing value at week 48 including those who did not enter RA-BEYOND. LE, linear extrapolation; LOCF, last observation carried forward; mITT, modified intent-to-treat; mTSS, modified Total Sharp Score; n, number of mITT patients; n, number of patients included in the analysis (with baseline and at least one postbaseline X-ray available for analysis); QD, once daily; RA, rheumatoid arthritis.
Efficacy
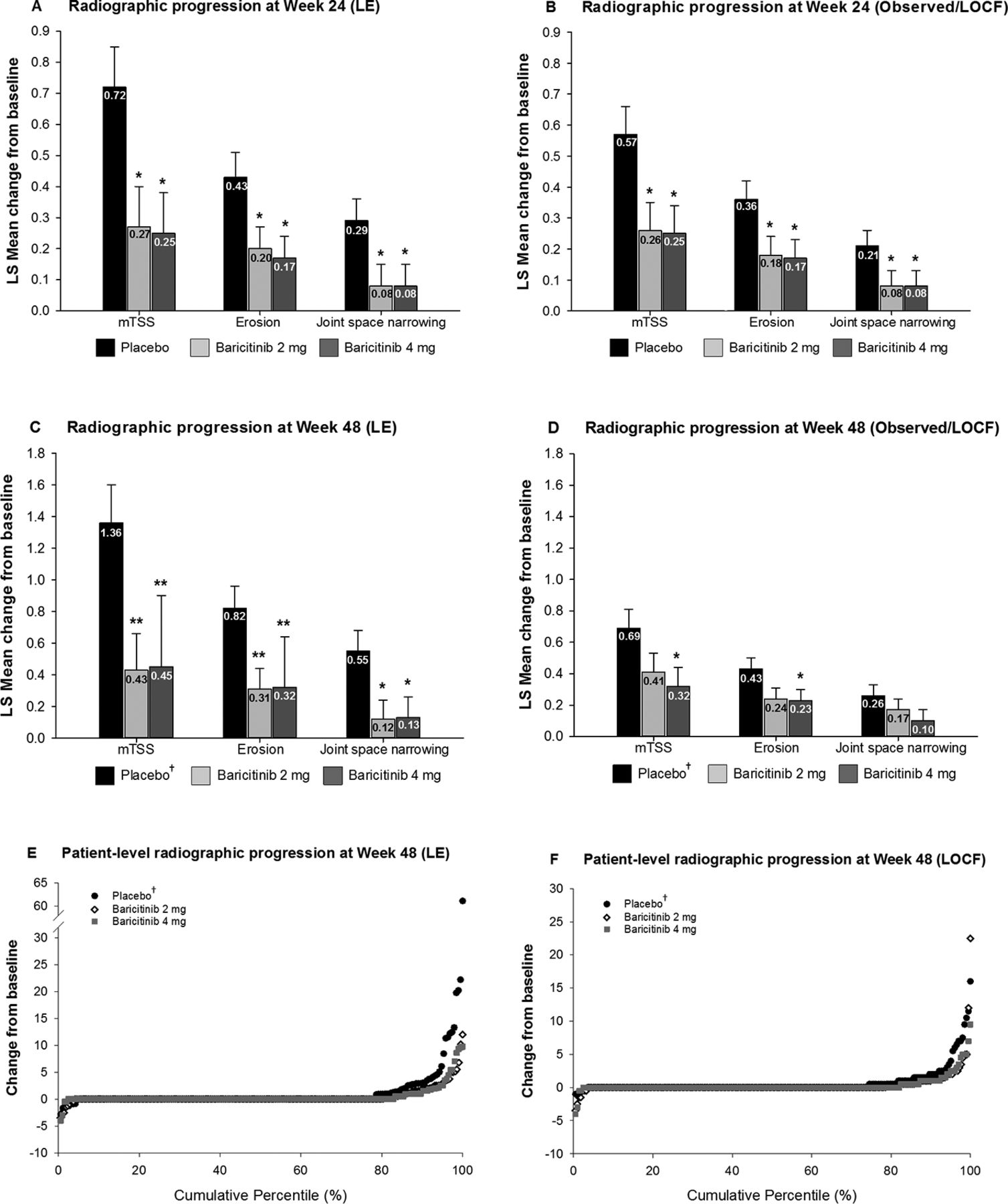
Using the primary LE analysis, compared with placebo, both doses of baricitinib inhibited progression of structural joint damage as measured by mTSS, and the subcomponents of joint space narrowing and bone erosion, at week 24. These differences persisted when analysed using the observed/LOCF method (figure 3A,B). Observations at week 48 revealed a similar pattern as the primary LE analysis. However, at week 48, the observed/LOCF method of analysis demonstrated that compared with patients initially randomised to placebo, only baricitinib 4 mg demonstrated statistically significantly less radiographic progression (including for mTSS and bone erosions (figure 3C,D)). Figure 3E and F presents individual patient data on the progression at week 48 for both the LE and LOCF methods in probability plots. At week 24, compared with placebo, a numerically larger proportion of patients had no progression in mTSS (ie, changes exceeding 0, 0.5 Sharp units or the SDC) for both the baricitinib 2 and 4 mg groups using both LE and observed/LOCF analyses (online supplementary table S1). Compared with placebo, the difference was statistically significant only for the 4 mg dose and based on the SDC threshold. Observations at week 48 were consistent with those at week 24 (table 1).
Supplementary file 1
Proportion of patients with no radiographic progression at week 48

{kind=link}
{kind=link}
{kind=link}
Inhibition of radiographic progression of structural joint damage at weeks 24 and 48. (Panels A–D) The LSM change from baseline in structural joint damage evaluated using mTSS, joint space narrowing and erosion score. (Panels E and F)Change from baseline in structural joint damage evaluated using the cumulative percentile change in mTSS. *P≤0.05, **P≤0.01, ***P≤0.001 versus placebo. †These patients were initially randomised to placebo but switched to baricitinib 4 mg at rescue or at week 24 prior to entry to RA-BEYOND. LSM, least squares mean; mTSS, modified Total Sharp Score.
Safety
Safety results for the 24-week placebo-controlled trial RA-BUILD have been previously published.8 Briefly, during weeks 0–24, the rate of treatment-emergent adverse events (TEAEs), serious adverse events (SAEs) (including serious infections) and discontinuations were similar across placebo, baricitinib 2 mg and baricitinib 4 mg groups (table 2); herpes zoster was reported in both baricitinib groups, but not in placebo. During weeks 0–48 (RA-BEYOND), both baricitinib dose groups reported 8% discontinuations, with more TEAEs and SAEs (including serious infections) in the baricitinib 4 mg group.
AE overview
Discussion
Treatment with baricitinib 2 or 4 mg once daily was associated with reduced rates of structural progression, measured using mTSS, through 1 year in patients with RA who had an inadequate response to csDMARDs. Both 2 and 4 mg doses of baricitinib were associated with structural benefit, but the 4 mg dose demonstrated more consistent reduction of structural progression when examined using both the LE and observed/LOCF analysis methods at both timepoints. The effect was seen for both the erosion and the joint space narrowing score. All radiographs, including baseline and week 24, were re-read as part of the scoring of the week 48 data. The change in mTSS at week 24 is consistent with the original reductions in structural progression reported in Dougados et al 2017.8 Additionally, patients initially randomised to placebo who then switched to active treatment after 24 weeks had a larger increase in mTSS at the 48-week timepoint than patients who were initially randomised to treatment with baricitinib. This is consistent with previous findings that earlier control of disease is associated with reduced radiographic progression.
Treatment with baricitinib not only reduced the rate of radiographic progression at week 48, but clinical efficacy (SDAI and HAQ-DI) was also maintained out to week 48 (online supplementary table S2).12 No new safety signals appeared through week 48. Rates of SAEs (including infections) and TEAEs were higher in the baricitinib 4 mg group than the baricitinib 2 mg group during weeks 0–48, while AEs leading to discontinuation were similar across baricitinib groups at week 48.
These findings are consistent with those seen in other studies evaluating the effects of treatment with baricitinib on structural progression over 1 year.5 6 For a molecule to be classified as a DMARD, an ability to inhibit the rate of radiographic progression of structural joint damage must be shown.4 These data provide further support that baricitinib is an effective DMARD for treating RA in csDMARD-inadequate response patients. Data from an open label LTE describing the long-term effects of tofacitinib on progression of radiographic structural damage were recently reported.13 Slightly over 1000 patients were followed for up to 48 months; the patients were receiving either 5 or 10 mg two times per day of tofacitinib with and without background csDMARDs. Mean change in mTSS from entry into the LTE through 12 months was 0.25 for all patients. Although it is dangerous to compare data across trials, the baricitinib data reported in patients who all had an inadequate response to csDMARDS were similar.
This study had limitations. Patients who completed the RA-BUILD study were included in this analysis whether or not they entered the long-term extension study, RA-BEYOND; however, 85% of the randomised and 95% of the completed patients of RA-BUILD entered RA-BEYOND, thereby limiting the influence of dropouts. Data were imputed for patients who did not enter the extension study or those with missing radiographs. The extension study was open-label in that all patients were aware that they were receiving baricitinib. However, the original treatment assignment as well as the actual dose of baricitinib remained blinded to the patient and investigator, and the readers scored all images blinded to all clinical information. Finally, the focus of the present manuscript is radiographic progression of joint damage from a single Phase three study and associated long-term extension data - limited conclusions can be drawn from the safety analyses, as evaluations from larger datasets that integrate information across multiple studies are better suited for this purpose.14
In summary, these data provide evidence that the positive structural efficacy response in csDMARD-IR patients initially observed at 24 weeks was maintained over 48 weeks of treatment with baricitinib. The most robust benefit across measures of radiographic progression was seen with the 4 mg dose. Additional annual radiographic assessments will continue during the extension study to determine if these affects are maintained during longer term treatment.
Acknowledgments
The authors would like to thank the patients who participated in this study. They also thank Stephanie Colvin, PhD, employee of Eli Lilly and Company, for creating the tables and figures and for assisting with manuscript preparation and process support.
References
Footnotes
Contributors All authors participated in the analyses and interpretation of data, provided critical comments and input, and reviewed and approved the final manuscript.
Funding This study was funded by Eli Lilly and Company and Incyte Corporation.
Competing interests DvdH has received consulting fees from AbbVie, Amgen, Astellas, AstraZeneca, BMS, Boehringer Ingelheim, Celgene, Daiichi, Eli-Lilly, Galapagos, Gilead, Janssen, Merck, Novartis, Pfizer, Regeneron, Roche, Sanofi, UCB and is director of Imaging Rheumatology bv. MD has received grant/research support or consulting support from AbbVie, Bristol Myers Squibb, Eli Lilly and Company, Novartis, Pfizer, Roche, Sanofi, and UCB. PE has received grant/research support or consulting support from Abbott, AbbVie, Bristol Myers Squibb, Eli Lilly and Company, MSD, Novartis, Pfizer, Roche, Samsung, Takeda and UCB. Y-CC has received speakers bureau fees and/or grant research support from AbbVie, Bristol Myers Squibb, Eli Lilly and Company and Pfizer. MG received research support from Eli Lilly and Company. RK is an employee of IQVIA. LX, SLW, IdlT, TPR, DES and SdB are employees of Eli Lilly and Company and may own stock or stock options in Eli Lilly and Company.
Ethics approval Institutional Review Board or Ethics Committee for each center.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Lilly provides access to relevant anonymised patient level data from studies on approved medicines and indications as defined by the sponsor specific information.