
Abstract
Osteoarthritis (OA) is one of the most common forms of degenerative joint disease and a major cause of pain and disability affecting the aging population. It is estimated that more than 20 million Americans and 35 to 40 million Europeans suffer from OA. Analgesics and non-steroidal anti-inflammatory drugs (NSAIDs) are the only therapeutic treatment options for OA. Effective pharmacotherapy for OA, capable of restoring the original structure and function of damaged cartilage and other synovial tissue, is urgently needed, and research into such disease-modifying osteoarthritis drugs (DMOADs) is in progress. This is the first of three reviews focusing on OA therapeutics. This paper provides an overview of current research into potential structure-modifying drugs and more appropriately targeted pharmacological therapy. The challenges and opportunities in this area of research and development are reviewed, covering the most up-to-date initiatives, trends, and topics.
Similar content being viewed by others

Introduction
The ever-expanding aging population expects and deserves a fulfilling and active life, with low dependence on “managed care”. This requires a healthy and well-functioning musculoskeletal system. However, age-related musculoskeletal diseases are a major cause of global morbidity, and result in high costs for health and social care systems. Chronic and inflammatory joint disease is a major cause of disability for the elderly. As life expectancy increases the incidence of musculoskeletal disease will grow, meaning there is an acute need for intervention to prevent and treat these diseases.
Advancing age is a major risk factor for degenerative joint disease. Osteoarthritis (OA) is a disabling degenerative joint disease that affects more adults than any other rheumatic disease does, and is the main cause of pain and disability among the elderly. Prevalence of OA increases with age: the disease affects 10 % of males and 18 % of females over 45, and these figures are predicted to rise as the general population ages. OA causes joint pain, stiffness, and loss of function, predominantly affecting the knee, hip, hand, spine, and other weight-bearing joints. A 2005 study in the USA estimated that OA is one of the five main causes of disability for non-hospitalized adults (source: Centers for Disease Control and Prevention (CDC, http://www.cdc.gov/), USA). The National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS, http://www.niams.nih.gov/) estimates that more than 20 million Americans suffer from OA. It is expected that by 2030, 20 % of adults in Western Europe and North America will have OA. Therefore, OA is expected to be a heavy economic burden on healthcare systems and community services in Europe and the rest of the world as the population expands and the number of elderly people increases.
OA is characterized by degeneration of articular cartilage, low-grade synovial inflammation (synovitis), and alterations to peri-articular and subchondral bone [1]. Traditionally, OA has been regarded as a “wear and tear” degenerative joint condition. However, recent studies have revealed that systemic factors regulate the metabolism of joint tissue, and that substantial cross-talk occurs between different joint tissues [2]. This means OA affects the whole joint, including cartilage, subchondral bone, synovium, tendon, and muscle [3, 4•, 5, 6].
As described earlier, OA is primarily associated with aging. However there are other important contributing factors, including obesity (which increases mechanical stress), history of joint trauma or repetitive joint use, genetics, inherited and acquired metabolic disorders, muscle weakness, underlying anatomical and orthopedic disorders (e.g. congenital hip dislocation), joint infection, crystal deposition, previous rheumatoid arthritis, and a variety of bone turnover and blood clotting disorders. There is increasing evidence for a connection between metabolic dysfunction and OA [2, 7]; indeed, metabolic osteoarthritis has recently been described as a subtype of OA [7].
Prevalence of OA is significantly higher in women, especially postmenopausal women and women with co-morbid metabolic bone conditions including osteoporosis (OP) [7]. Although the underlying causes of women’s increased susceptibility to OA are not fully understood, research is beginning to focus on associations with sex hormones, obesity, and physical activity to determine whether modifiable factors including estrogen, weight management, and protection during sport and exercise can be used as treatment for postmenopausal women with OA and OP [2, 8].
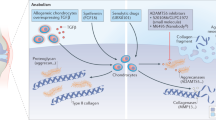
OA has an important inflammatory component that includes increased activity of several cytokines and chemokines in the joints [9•]. These inflammatory cytokines and chemokines drive the production and secretion of enzymes that mediate destruction of cartilage [1]. Cartilage aging drives cellular alterations that result in a damage-induced, senescence-associated secretory phenotype characterized by production and secretion of cytokines, chemokines, and proteases [10, 11•]. Oxidative stress and inappropriate mechanical signals can further promote the senescence-associated secretory phenotype of aging chondrocytes [10, 12], as observed in tumor cells [13••].
OA is now regarded as a disease of the whole joint, and this must be taken into consideration when evaluating new and old treatments [14]. Our understanding of the pathogenesis of OA is rapidly increasing and is expected to assist development of disease-modifying therapy. However, very little new pharmacological therapy has been introduced and use of existing symptom-modifying drugs with deleterious side effects continues. Limitations of conventional medical management of OA indicate the need for novel, safe and effective treatment for OA patients. Current therapy insufficiently addresses clinical need, and there is no effective pharmacological or biological therapy capable of restoring the original structure and function of damaged cartilage and other synovial tissue in OA or any other form of arthritis. This is the first of three reviews of recent progress in OA therapeutics. This paper will address the topic of targeted pharmacological therapy.
Existing Pharmacotherapy for OA
Current pharmacological intervention that addresses chronic pain in OA is insufficient, and no proven structure-modifying therapy is available [1]. The main clinical guidelines recommend use of mild analgesics, for example acetaminophen (paracetamol), for treatment of mild-to-moderate OA symptoms, and only recommend use of nonsteroidal anti-inflammatory drugs (NSAIDs) after acetaminophen has failed [15]. NSAIDs are the most common treatment for rheumatological conditions including OA, rheumatoid arthritis (RA) and gout. These drugs are extensively used throughout the world and have analgesic, antipyretic and, at higher doses, anti-inflammatory effects. In the UK, almost a quarter of patients visiting their general practitioner for “arthritic” and “rheumatic” complaints are prescribed NSAIDs. The trend is similar for other industrialized nations. NSAIDs relieve pain and increase mobility for approximately 60 % of patients with OA, acting by inhibiting activity of the cyclooxygenase (COX) enzymes [16]. There are two isoforms of COX: COX-1, which is constitutively expressed, and COX-2, which is an inducible isoform. COX-2 expression is normally low, but is increased by inflammatory stimuli and cytokines [16]. Animal studies and data from humans reveal that COX-2 up-regulation in OA and RA is associated with pain and inflammation [17]. Prostaglandins synthesized by the constitutively-expressed COX-1 have “cytoprotective” functions in the stomach and are involved in maintaining normal physiological function of the stomach lining. Inhibition of the COX-1 isoform affects these important homeostatic and gastroprotective functions, and has substantial consequences for gastrointestinal health [18]. NSAIDs have a wide variety of side effects, but the most clinically important are upper gastrointestinal tract dyspepsia, peptic ulceration, hemorrhage, and perforation, leading to death for some patients. Most conventional NSAIDs inhibit both COX isoforms. However, some NSAIDs, including meloxicam [19] and celecoxib, are more selective towards COX-2 [17]. COX-2-selective inhibitors provide effective pain relief for patients with OA and RA, and have similar efficacy to traditional NSAIDs. However, cost-effectiveness and cost-utility studies suggest that their use should be limited to patients with severe upper gastrointestinal side effects and ulcers [20]. For patients with knee OA, aceclofenac reduces pain, reduces disease severity, and improves functional capacity to a similar extent to diclofenac, piroxicam, and naproxen [21]. As with traditional NSAIDs, nephrotoxicity and hypertension are concerns with COX-2 inhibitors [22]. Despite their side effects, NSAIDs are recommended by the European League Against Rheumatism (EULAR, http://www.eular.org/), and the Osteoarthritis Research Society International (OARSI, http://www.oarsi.org/) treatment guidelines committee recommends NSAIDs for management of hip and knee OA [23]. More selective pharmacotherapy is needed for treatment of OA and related arthritic conditions.
Should Cartilage or Subchondral Bone be Targeted?
Articular cartilage in load-bearing joints requires a solid and stable foundation of subchondral bone. There is ongoing debate about the function of subchondral bone in OA [3, 24–26, 27•]. Although many researchers support the idea that OA is a disease of articular cartilage, there is increasing support for the theory that subchondral bone should be a priority target of OA treatment [26]. It is believed that vascular pathology and loss of mineral density in subchondral bone are important to initiation and/or progression of OA [25], and that changes to subchondral bone may accelerate progression of pre-existing disease [28]. Several recent studies have emphasized the importance of the cartilage–bone interface in OA, observing that cartilage and subchondral bone act as a single functional unit in health and in disease, and that alteration of either tissue, resulting from injury or abnormal loading, may alter the biomechanical status of the other. This concept has recently been discussed in detail by other investigators [3, 24]. Other papers have questioned whether there is any compelling evidence to suggest that OA can be modified, and whether the current treatment focus on articular cartilage is appropriate [29, 30]. Synovitis and abnormal subchondral bone turnover also contribute to disease progression, and are associated with OA pain [33]. Subchondral bone is an attractive target for disease-modifying osteoarthritic drugs (DMOADs) [26]; it is increasingly recognized that OA is a disease of the whole joint [14, 31], and that the bone–cartilage unit is especially important [32•].
Bisphophonates and Strontium Ranelate—Linking Osteoporosis Treatments to OA
Bone remodeling is a continuous process of old bone resorption and new bone formation. It is a natural, physiologically regulated process, occurring during growth, development, and adaptation to mechanical load and physical exercise. Bone remodeling controls the reshaping and replacement of bone after traumatic injury, for example fracture, and after micro-damage, which occurs during intensive physical activity. In coordination with endocrine signals, it responds to the functional demands of mechanical loading. Imbalanced regulation of bone resorption and bone formation results in metabolic bone disease, including osteoporosis (OP) [34]. OP is a bone disease that causes bones to become more porous, resulting in reduced bone mineral density and bone mass. This gradually makes bones weaker, more brittle, and more fragile, with long bones becoming substantially more prone to fracture. OP results from an imbalance in the bone remodeling process whereby bone resorption, which is mediated by osteoclasts, outpaces bone formation, which is mediated by osteoblasts. It can also occur as a consequence of chronic joint disease: for example, subchondral sclerosis is associated with age-related joint degeneration [35].
The rationale for targeting bone when treating OA is based on molecular cross-talk between subchondral bone and cartilage [32•], and the subchondral bone resorption that occurs at an early stage in development of OA [46]. Abnormalities of subchondral bone metabolism, especially increased bone turnover, have been detected in the early stages of some forms of OA [26]. If osteoclastic bone resorption reduces bony support for the overlying cartilage, this can facilitate progression of OA [25]. Studies of people with knee OA have observed that cartilage loss and risk of knee replacement are higher if subchondral bone cysts are present than if only bone marrow lesions (BMLs) are present, suggesting that cysts identify those most likely to benefit from prevention of disease progression [36]. These studies support the hypothesis that subchondral bone is important to progression of joint disease, and that factors which result in cartilage destruction may come from subchondral bone [37]. Therefore, strategies for new treatment to inhibit progression of OA must consider the subchondral bone compartment [25, 37]. In OP and OA, both high and low bone mass conditions may result in disease induction and/or progression [26].
Substantial progress has been made over the last five decades in non-hormonal OP treatment [38]. Bisphosphonates (BPs) are a class of drugs used to treat OP and related bone disease by preventing loss of bone mass. They were developed in the early 1960s as a potential treatment for bone disease. BPs are the most frequently prescribed drug for treating osteoporosis and other diseases characterized by increased bone resorption. For patients with postmenopausal OP, BPs reduce osteoclast activity to healthy, pre-menopausal levels, reducing the rate of bone loss. BPs increase bone mass, strengthen bones, and reduce incidence of fracture, including severe fractures of the hip and spine. BPs approved for treatment and/or prevention of osteoporosis include alendronate (Fosamax, Fosamax Plus D; Merck), ibandronate (Boniva; Genentech), zoledronic acid (Reclast; Novartis), and risedronate (Actonel, Actonel with Calcium, and Atelvia; Warner Chilcott). Other BPs include etidronate and raloxifene, used for secondary prevention of osteoporotic fragility fractures in postmenopausal women. In addition to treating OP, BPs are also used to reduce calcium levels in the blood and to treat Paget’s disease of bone (which causes bones to become weak and deformed) and bone-related cancer, alleviating pain and weakness. BPs are also used after other forms of cancer treatment, including chemotherapy and hormone therapy, both of which can weaken bone. They can also prevent some cancers spreading to bone. BPs are an extremely important class of drugs and, in addition to their therapeutic benefits for OP and related bone disease, are potential therapeutic agents for disease modification in OA [39]. They may also lead to new techniques for investigating the pathogenic mechanisms relating synovitis [4•], subchondral bone pathology [25, 26], and OA pain [33].
However, it is important to note that mixed results have been obtained from clinical trials of risedronate. The effect of risedronate on joint structure and symptoms of knee OA was studied in the BRISK randomized, controlled trial [40]. The study was a one-year prospective, double blind, placebo-controlled study that enrolled patients (40–80 years of age) with mild to moderate OA of the medial compartment of the knee. The study concluded that significant improvements of joint structure and symptoms were observed for patients with primary knee OA treated with risedronate [40]. A subsequent multinational trial failed to find any effect on structural outcomes. The knee OA structural arthritis (KOSTAR) study tested the efficacy of risedronate for providing symptom relief and slowing disease progression for patients with knee OA [41]. This study found that although risedronate reduces biochemical markers of cartilage degradation, it does not reduce symptoms or slow radiographic progression for patients with medial compartment OA of the knee. Although these UK-based studies reported some favorable effects for risedronate compared with placebo, the cohorts selected included too few cases of progressive radiographic OA to demonstrate chondroprotection [42]. Early animal studies indicated that risedronate should work, as the drug can reduce bone marrow lesions. However, the animal data did not transfer well to human subjects. The patients were not properly stratified, and this could be one reason the trials failed.
Strontium ranelate, a strontium salt of ranelic acid, is a drug for treating osteoporosis (OP); it is marketed as Protelos or Protos by the French pharmaceutical company Servier (http://www.servier.com/). Ranelic acid (molecular formula: C12H10N2O8S) is an organic acid that chelates metal cations (Fig. 1). In Europe strontium ranelate has been authorized for treatment of osteoporosis in postmenopausal women to reduce the risk of vertebral and hip fracture. Strontium ranelate is a bone density conservation agent—these drugs inhibit bone resorption and favor bone extracellular matrix formation and mineralization. They are used to heal fractures and to treat metabolic bone diseases, including OP. The drug is unusual in that it both increases deposition of new bone by osteoblasts and reduces resorption of bone by osteoclasts. It is therefore promoted as a “dual action bone agent” (DABA). Strontium ranelate is registered in many countries as a prescription drug to reduce the risk of vertebral and hip fracture associated with postmenopausal OP. In the UK, strontium ranelate is prescribed under the National Health Service (NHS, http://www.nhs.uk/) for treatment of postmenopausal OP. In addition to the “fragility fracture risk assessment” pathway (http://pathways.nice.org.uk/pathways/osteoporosis/osteoporosis-overview), the National Institute for Health and Clinical Excellence (NICE, http://www.nice.org.uk/) has published updated final guidelines for use of alendronate, etidronate, risedronate, raloxifene, strontium ranelate, and teriparatide in England and Wales for prevention and treatment of osteoporotic fragility fractures in postmenopausal women (http://www.nice.org.uk/nicemedia/live/11748/42472/42472.pdf). In the USA, strontium ranelate is not approved for treating OP by the US Food and Drug Administration (FDA, http://www.fda.gov/) because of safety concerns and reports of increased risk of blood clots and memory loss. Studies suggest that patients receiving 2 g day−1 strontium for 3–4 years are at greater risk of vascular side effects (i.e. blood clots) and nervous system side effects (seizures). Rizzoli and colleagues recently reviewed drug–drug interactions for OP patients. These are relatively rare and although there are risks, these should be weighed against the benefits of treatment [43].

Molecular structure of strontium ranelate, also known as: Protelos, 135459-87-9, Osseor, Protos, Ranelic acid distrontium salt. Molecular formula C12H10N2O8SSr2 (molecular weight 517.5214)
A systematic review published in 2006 reviewed the literature on strontium ranelate to determine its efficacy at preventing and treating postmenopausal OP [44]. The authors searched the MEDLINE, EMBASE and Cochrane Library databases from 1996–2005. They also included relevant conference proceedings from the previous two years. The review included randomized controlled trials (RCTs) of at least one-year duration comparing strontium ranelate with placebo. Four trials were included in the review, which provided silver-level evidence (a term introduced by the Cochrane Musculoskeletal Group and explained at http://musculoskeletal.cochrane.org/) for the efficacy of strontium ranelate for both fracture reduction for postmenopausal women with OP and increasing BMD for postmenopausal women with or without OP.
A recent clinical trial indicated that strontium ranelate can slow progression of knee OA, making it the first trial to achieve a positive result when investigating a drug with potential as a disease-modifying osteoarthritic drug (DMOAD). The “strontium ranelate efficacy in knee osteoarthritis trial,” or SEKOIA, published in 2012, suggested the drug can significantly affect progression of knee OA compared with placebo. In a double-blind randomized controlled trial, strontium ranelate reduced knee OA pain symptoms, improved function, and reduced radiography-detectable cartilage loss, as indicated by reductions in joint-space narrowing over three years [45•]. The trial was a phase III, multicenter, international, double-blinded, placebo-controlled study of men and women 50 years of age or above with a clinical diagnosis of knee OA as defined by the American College of Rheumatology (ACR, http://www.rheumatology.org/). The investigating team’s objective was to evaluate the efficacy and safety of two doses of strontium ranelate (1 g and 2 g day−1) versus placebo, administered orally over three years, for treating people with knee OA. The primary endpoint of the study was radiographic progression of knee OA, and assessment of joint space narrowing (JSN) determined by use of radiography. The main secondary endpoints for the study were the Western Ontario and McMaster Universities osteoarthritis index (WOMAC, http://www.rheumatology.org/practice/clinical/clinicianresearchers/outcomes-instrumentation/WOMAC.asp), used to measure pain functional assessment of the target knee, and knee pain intensity assessed during the 48 h before the visit by use of a visual analogue scale. Treatment with strontium ranelate was associated with reduced progression of cartilage degradation. Administration of strontium ranelate (2 g day−1) significantly reduced total WOMAC score and pain subscore and revealed a possible trend for improved physical function. The study concluded that treatment with strontium ranelate is associated with significant beneficial effects on joint structure for patients with knee OA when the drug is administered at 2 g day−1.
According to the Medicines and Healthcare products Regulatory Agency (MHRA), an executive agency of the Department of Health in the UK responsible for ensuring that medicines and medical devices are effective and acceptably safe, strontium ranelate is not recommended for patients with current or previous venous thromboembolism (VTE) or for patients who are temporarily or permanently immobilized (e.g. post-surgical recovery or prolonged bed rest) and thus at increased risk of VTE (http://www.mhra.gov.uk/Safetyinformation/DrugSafetyUpdate/CON152727). The need for continued treatment with strontium ranelate should also be re-evaluated for patients over 80 years old who are at risk of VTE. Strontium ranelate is also associated with serious skin and hypersensitivity reactions, including drug rash with eosinophilia and systemic symptoms (DRESS). In addition, rare serious skin reactions may occur during the first weeks of treatment.
The European Medicines Agency (EMA, http://www.ema.europa.eu/) has completed a review of strontium ranelate. Although a positive benefit–risk balance has been confirmed for the drug, new contraindications and revised warnings have been issued (http://www.ema.europa.eu/docs/en_GB/document_library/Press_release/2012/03/WC500124206.pdf). The EMA Committee has concluded that these medicines are an important treatment for women with osteoporosis, but changes to prescribing advice are necessary to better manage associated risks. A European Commission decision on this opinion is expected in the near future.
Further clinical studies on the efficacy and safety of bisphosphonates are required to determine whether this drug is capable of disease modification in OA. Clinical trials on cohorts of younger OA patients are needed to confirm the drug’s effects and assess its safety.
Inhibitors of Inducible Nitric Oxide Synthase (iNOS)
Nitric oxide (NO) gas has important biological properties. NO is a signaling molecule involved in many physiological and pathological processes. It was initially identified as endothelium-derived relaxing factor (EDRF) by Robert F. Furchgott, Louis J. Ignarro, and Ferid Murad, who shared the 1998 Nobel Prize in Physiology or Medicine for its discovery. NO is a free radical synthesized from l-arginine by the NO synthase enzymes (NOS). It is now well established that production of NO by iNOS is stimulated by cytokines, including IL-1β, interferon-γ (IFN-γ), and tumor necrosis factor-α (TNF-α). These mediators induce iNOS expression in a variety of cells, including chondrocytes, synoviocytes and macrophages. NO production is increased in inflammatory arthritides. Increased NO production is observed in a variety of compartments in vivo, but inflammatory synovium and cartilage are the main sources of NO in the joint. Catabolic and proinflammatory cytokines, NO, prostaglandin E(2) (PGE2) [5], and neuropeptides [47], produced by the inflamed synovium in OA [4•], collectively alter the balance of cartilage matrix degradation and repair, eventually leading to excess production of the proteolytic enzymes responsible for cartilage breakdown [48]. Because synovitis is associated with clinical symptoms of OA and reflects joint degradation in OA, it has been suggested that synovium-targeted therapy could help alleviate symptoms and prevent structural progression [4•].
Induced NO, in addition to being a “final common mediator” of inflammation, is essential for induction, up-regulation, and amplification of inflammatory response [49]. NO is believed to be involved in cartilage inflammation and catabolism and the pain associated with OA [50], and NO and iNOS are therefore regarded as potential targets for therapeutic intervention [51]. Onset of arthritis in rodent models has been successfully blocked by the NOS inhibitor NG-monomethyl-l-arginine (L-NMMA) [51], suggesting possible involvement of NO in OA pathogenesis and tissue destruction.
Diacerhein, also known as diacetylrhein, is an anti-inflammatory drug used in OA treatment. It works by blocking the action of IL-1β, and is a potent inhibitor of IL-1β-induced NO production by chondrocytes [52]. A Cochrane Database systematic review published in 2006 suggested that diacerein leads to a small, but consistent, improvement of OA pain [53]. The European Medicines Agency (EMA, http://www.ema.europa.eu/ema/) has initiated a review (http://www.ema.europa.eu/docs/en_GB/document_library/Referrals_document/Diacerein/Procedure_started/WC500135576.pdf) of diacerein-containing medicines used to treat the symptoms of OA and other joint diseases. This action is in response to evidence from clinical trials and scientific literature suggesting that the efficacy of diacerein against OA is weak. The EMA will review the data on the benefits and risks of diacerein and issue an opinion on the marketing authorization of diacerein-containing medicines across the European Union.
Emerging evidence suggests that NO and its redox derivatives may also have protective functions in the joint [50]. For example, in culture models of chondrocytes, addition of exogenous NO inhibits proinflammatory activation by preventing nuclear localization of the transcription factor nuclear factor-κB (NF-κB). In contrast, the presence of peroxynitrite, a redox derivative of NO, increases the inflammatory response of chondrocytes by sustaining nuclear localization of NF-κB [50]. Additionally, under some conditions, exogenous NO can stimulate collagen synthesis in cultured rat fibroblasts and human tendon cells. The protective functions of NO for multiple cell types, with the opposing activity in cultured chondrocytes suggest that NO may have additional protective effects for chondrocyte function.
Metabolic Targets: Mitochondria
Mitochondrial diseases are believed to be caused by the failure of mitochondria. Mitochondria dysfunction has been implicated in age-related diseases and the aging process [54]. Chondrocytes age as the extracellular matrix of cartilage undergoes age-dependent changes. Consequently, the biosynthetic activity of chondrocytes declines, and their responsiveness to anabolic mechanical stimuli and growth factors decreases [55]. There is increasing interest in the effect of mitochondrial abnormalities associated with aging and mitochondrial dysfunction on OA [56, 57]. Degradation of mitochondrial DNA and loss of the mitochondrial membrane potential may affect pathogenesis of OA, especially because respiration-mediated ATP production and mitochondrial function are sensitive to nitric oxide (NO) and are vital for matrix synthesis [58].
Mitochondrial functions, including mitochondrial respiratory chain activity and ATP synthesis, have been observed to be altered in OA chondrocytes [59•]. Francisco Blanco’s research team revealed that respiratory activity of OA chondrocytes is altered as a result of the reduced activity of mitochondrial complexes II and III [60].
Somatic mutations of mitochondrial DNA (mtDNA) have been described in synoviocytes from RA joints [61]. Studies of chondrocytes from OA joints suggest there is also mitochondrial dysfunction in OA and, as for RA, this may originate from somatic mutations in mtDNA [62, 63]. Polymorphisms in mtDNA could be useful biomarkers for the diagnosis and prognosis of OA [59•]. Studies are in progress to determine whether these mutations induce inflammatory damage in RA and OA, or whether they are epiphenomena of cellular damage induced by the proinflammatory cytokines, prostaglandins, reactive oxygen species (ROS), and nitric oxide (NO) of chronic inflammatory joint disease. It is known that proinflammatory cytokines, including TNF-α and IL-1β, regulate mitochondrial function in human articular chondrocytes by reducing the activity of the mitochondrial respiratory chain and reducing ATP levels [55]. The mitochondrion is strongly affected by proinflammatory cytokine-mediated toxicity in chondrocytes [63]: progressive ATP depletion caused by NO release in chondrocytes reduces mitochondrial reserves [64]. This NO-mediated mitochondrial dysfunction [55] suggests that targeting NO production and iNOS might support mitochondrial function in chondrocytes. Maintaining mitochondrial DNA integrity is an important potential preventive measure that might protect bioenergetic processes in chondrocytes from the effects of NO, IL-1β and TNF-α [63]. However, it is difficult to conceive how mitochondria could be targeted for therapeutic purposes.
ROS, for example superoxide, hydrogen peroxide, and hydroxyl radicals, are usually produced in mitochondria when electrons leak from the electron-transport chain and react with oxygen to form superoxide. ROS levels are controlled via multiple enzyme systems, including superoxide dismutase (SOD), catalase, glutathione S-transferase and thioredoxin. Production of hydrogen peroxide (H2O2) by inflammatory and synovial cells is an important cause of cellular damage during joint inflammation. Hydrogen peroxide is formed by dismutation of superoxide and by oxidases. SOD converts superoxide to hydrogen peroxide, which is then removed by glutathione peroxidase or catalase, preventing formation of such highly aggressive ROS as peroxynitrite or the hydroxyl radical [65]. Living cells maintain a complex and interrelated protective system, involving endogenous antioxidant vitamins, use of minerals including selenium and manganese as cofactors, and glutathione, to protect themselves from the harmful effects of ROS [66, 67]. Chondrocytes use a variety of antioxidant enzymes, including catalase, SOD and a variety of peroxidases, to reduce and control cellular levels of ROS. Recent work from David Young’s group has revealed that SOD is downregulated during OA progression and in end-stage disease [68]. Depletion of SOD in chondrocytes increases ROS levels [68]. This work confirms observations made by Francisco Blanco’s group several years earlier [69]. Ruiz-Romero et al. [69] used a proteomics approach, based on two-dimensional DIGE and MALDI-TOF–TOF mass spectrometric identification of mitochondria-enriched protein fractions, to reveal a “redox imbalance” and reduced mitochondrial SOD levels in chondrocytes from OA cartilage.
In summary, a decline in mitochondrial function is believed to be important to the aging process and to aging-associated disease [54]. Mitochondrial dysfunction and DNA damage are implicated in the pathogenesis of OA [56, 59•, 63]. As cartilage ages, the cells and their organelles also undergo age-related changes and the number of chondrocytes declines [70, 71]. Mitochondrial dysfunction increases the inflammatory response to cytokines [72], induces COX-2 expression (at both mRNA and protein levels) [73] and is accompanied by oxidative stress, chondrocyte apoptosis, cytokine-induced chondrocyte inflammation and matrix catabolism, and chondrocalcinosis [58, 59•, 64]. Multiple factors are believed to contribute to mitochondrial dysfunction in OA [74]. These include inhibition of mitochondrial biogenesis via suppression of important mitochondrial transcription factors. Risk of mitochondrial dysfunction is increased for individuals with specific mitochondrial DNA haplotypes [75] and by acquired mitochondrial DNA mutations [59•, 61]. These mitochondrial changes, combined with defective chondrocyte autophagy, result in increased ROS and reduced endogenous antioxidants, and promote inflammatory responses, abnormal gene expression, and cell death [74].
A sedentary lifestyle, associated with OA-induced physical inactivity, is associated with reduced mitochondrial function [57]. Therefore, from a metabolic perspective, physical activity and an active lifestyle are potentially important preventive measures against OA [57]. Development of drugs and identification of naturally occurring compounds capable of altering mitochondrial function could complement strategies to reduce cartilage degradation in OA [63]. Regulating chondrocyte metabolism, autophagy (an essential, homeostatic and protective mechanism by which cells degrade their own components), and apoptosis (programmed cell death) may be achieved via pharmacological and physiological modulation of sirtuins. Sirtunins are a family of seven NAD(+)-dependent deacetylases, activated by NAD(+) and the antioxidant phytochemical resveratrol [76•]. Resveratrol has been revealed to protect chondrocytes against oxidant injury and apoptosis via its effect on mitochondrial repolarization and ATP production [77]. The author has recently reviewed the potential benefits of resveratrol for enhancing chondrocyte function [78, 79]. Dietary supplementation with resveratrol and related antioxidant phytochemicals may be an important nutritional preventive strategy for OA, especially for people with compromised antioxidant systems.
Anti-MMP Therapy
Enzymatic breakdown of collagen in articular cartilage is mainly mediated by matrix metalloproteinases (MMPs) [48]. MMPs are extracellular proteinases involved in cleavage of ECM components [80] and of proteins on the cell surface and in pericellular regions [81]. The MMPs identified to date can be grouped by their substrate specificity [82], and include collagenases (MMP-1, MMP-8, MMP-13, MMP-18), gelatinases (MMP-2, MMP-9), stromelysins (MMP-3, MMP-10, MMP-11), matrilysins (MMP-7, MMP-26), membrane type-MMPs (the MT-MMPs: MMP-14, MMP-15, MMP-16, MMP-17, MMP-24 and MMP-25), and several others (MMP-12, MMP-19, MMP-20, MMP-21, MMP-22, MMP-23, MMP-27 and MMP-28). More than 27 MMPs have been identified. Four of these, the collagenases MMP-1, MMP-8, and MMP-13, and the membrane-bound MMP-14, are important to cartilage collagen destruction [86]. MMP-13 has a substrate preference for type II collagen, and is believed to be the most important collagenase in cartilage [87]. It can also act as a gelatinase, enabling it to degrade collagen further than other collagenases do. The synthesis and activity of MMPs in cartilage and other joint tissue are tightly controlled by a variety of pro-inflammatory cytokines, growth factors, and tissue inhibitors. MMPs are secreted as inactive pro-enzymes and are usually activated by other proteinases. Inhibition occurs both locally (by tissue inhibitors of metalloproteinases (TIMPs)) and systemically (by α2-macroglobulin) [83–85]. In joint disease, increased expression of collagenolytic MMPs and reduced expression of their endogenous regulatory inhibitors, the TIMPs, disrupts the homeostatic balance between synthesis and destruction to favor collagen breakdown. This mechanism causes the extensive damage to the collagen fibrillar network observed in OA cartilage pathology which seems to be mediated primarily by the collagenases MMP-1 and MMP-13 [88].
The author has recently discussed MMP inhibitors in a review article published in Current Drug Targets [48]. Specific areas of interest within this topic are TIMPs, small molecule MMP inhibitors (MMPIs), antibody inhibitors (to be covered in greater detail in the second review in this series), anti-sense technology, and dietary phytochemical inhibition of MMPs (previously reviewed by the author in this journal [79]).
Mapp and co-workers studied the effects of M503902—an orally administered MMP inhibitor developed by AstraZeneca, with broad activity against a range of MMPs—on joint pathology, osteochondral angiogenesis, chondropathy, and pain in a rat meniscal transection (MNX) model of OA [89]. The authors found that treatment with the MMP inhibitor reduced weight-bearing asymmetry, attenuated chondropathy, and reduced vascularity. The authors propose that the association between osteochondral angiogenesis and pain may be explained by perivascular nerve growth or stimulation of subchondral nerves after loss of osteochondral integrity. The authors conclude that targeting angiogenesis may be useful for treatment of pain associated with OA structural damage.
Several herbal medicines have been investigated for anti-inflammatory and indirect MMP-inhibitory properties. These medicines include phytochemicals and flavonoids and catechins from green tea, rosehip, curcumin, and resveratrol (discussed in the section “Herbal therapy for OA” and reviewed in Ref. [48]).
The involvement of MMPs in normal physiological processes means that therapy involving total inhibition is clinically undesirable. However, partial inhibition of over-active MMPs may be beneficial when treating inflammatory conditions. Selective modulation to upregulate, activate, or downregulate targeted MMPs is a more appropriate objective. However, there has been no major new development in this area.
Herbal Therapy for OA
There is growing interest in herbal drugs and medicines, and the patient-driven search for alternative treatments and herbal medicines has intensified. There is substantial overlap between conventional and herbal medicine in treatment of arthritic, rheumatic, and musculoskeletal disease: some of the drugs in our pharmacopoeia (or their derivatives) have a long-established history in ethnopharmacology and have been used for centuries [79]. Detailed discussion of this topic is beyond the scope of this article. The author has recently reviewed this area of research in a review article published in Current Rheumatology Reports [79]. The author has also reviewed the potential prophylactic properties of plant-derived phytochemicals, including curcumin and resveratrol, and their use for targeting NF-κB signaling and inflammation in OA [78]. Available clinical data for resveratrol are encouraging and support its inclusion in future clinical trials [90]. Other researchers have reviewed the topic of herbal medicine and nutraceuticals for OA [91–96]. This area of research is constantly growing and expanding. Herbal remedies and botanical supplements have become important topics of research and clinical practice in orthopedics and rheumatology [94]. Many herbal medicines seem to be relatively safe, with some having an encouraging risk–benefit profile, and may offer a much-needed alternative for patients with OA [97]. Some herbal remedies are inhibitors of NF-κB, and may be able to reduce use of NSAIDs and the associated side effects. However, in-vivo data and large, randomized clinical trials to verify the theoretical reasons for using herbal medicine are largely lacking [79]. The author’s view is that herbal medicine could have benefits if used to supplement existing pharmaceutical treatment options [98] with the objective of reducing dosages and frequency of consumption of conventional drugs.
Lessons from Recent Setbacks and Failures
The ability to develop new drugs to slow progression of OA in the elderly population will have enormous public health implications [99]. However, the pharmaceutical industry is currently suffering from a productivity crisis, and there are very few new drugs approaching clinical readiness. Over the last decade the number of new compounds has decreased; however, more IND applications to the Food and Drug Administration (FDA) have been made over the last ten years than during the previous decade. This reveals the current state of the industry: the processes of discovery and preclinical development are highly productive, but very few compounds make it through the subsequent clinical evaluation phase. This trend has implications for the future of OA clinical trials and DMOAD development.
The pharmaceutical industry has had to learn from several failures and setbacks. The key points are summarized below:
-
Developing drugs that target enzymes involved in physiological, in developmental, and in pathological functions (e.g. MMPs) can be extremely challenging.
-
Biological approaches were largely ignored and regarded as the domain of small start-up biotechnology companies, many of which were acquired by larger drug companies in the last few years. Biological therapy will be covered in the next paper in this series.
-
It is difficult to test new drugs without sensitive biomarkers to help monitor patient response. The lack of such markers necessitates identification of a range of biochemicals that can be used to assess the efficacy of new formulations.
New Biomarkers to Stimulate Drug Discovery
A biomarker is a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacological responses to a therapeutic intervention [100]. OA is often diagnosed via radiography when clinical signs of pain and loss of mobility have already appeared. Unfortunately, despite being a leading technique, radiography has numerous weaknesses. It is an indirect measure of alterations in articular cartilage and does not measure a dynamic process. Changes observed over time are small, and occur in only a subset (progressors) of patients. Radiographs rarely correlate with symptoms and are poorly reproducible. By the time radiographic changes have been detected, disease has progressed extensively and cartilage degradation is quite substantial. OA is characterized by a prolonged pre-clinical “molecular” phase, a “pre-radiographic” phase, and a “recalcitrant radiographic” phase; by the latter phase there is already extensive structural change to joints, along with pain and loss of function. This is why more advanced imaging techniques, for example MRI, are more suitable for detecting early degenerative changes in the joint.
In 2011 the Osteoarthritis Research Society International (OARSI, http://www.oarsi.org/) and the US Food and Drug Administration (FDA, http://www.fda.gov/) established the OARSI FDA osteoarthritis biomarkers working group [101••]. This working group has divided OA biomarkers into two major groups: soluble or “wet” biomarkers and “dry” biomarkers [101••]. These biomarkers will facilitate earlier diagnosis and treatment. Linking a biomarker to a clinical endpoint can greatly assist the drug-discovery process. We have very few reliable biomarkers for OA, and more research is needed to identify markers that link to dynamic changes in the joint. In the future, biomarkers will be increasingly important in all the different phases of OA drug development, including regulatory review. However, only a small number of biomarkers are expected to become sufficiently accepted and established to serve in regulatory decision-making as surrogate endpoints substituted for traditional clinical endpoints [102]. Drug and biomarker development are interdependent, and future progress in OA drug discovery will require sensitive new biomarkers to predict OA progression and responses to new therapeutics.
Conclusions
There is an urgent need for new treatment options for patients suffering from OA. The pharmaceutical industry has tried and failed to provide effective and safe disease-modifying osteoarthritic drugs (DMOADs) for the millions of patients suffering from this debilitating disease [103]. Future therapeutics will probably require a combination of approaches. It seems unlikely that any single treatment will suffice for OA because of its complex etiology and multifactorial nature. Future therapy needs to be highly targeted and personalized, rather than the “one drug to treat all forms” model that has dominated thus far. Dietary and lifestyle intervention will need to be used alongside new DMOADs for treating OA as a global and systemic joint disease. In addition, OA pain needs to be addressed by use of a combinatorial approach or by beginning DMOAD treatment at a much earlier stage, before any symptoms have appeared. However, this will be extremely challenging to achieve because patients do not present before the occurrence of pain.
For decades OA has been viewed as a “wear and tear” disease leading to loss of cartilage. This concept is increasingly being challenged, as is the potentially misleading terminology—e.g. “osteoarthrosis”—used to describe the “wear and tear” [104••]. Instead, OA is increasingly viewed as a “low grade” inflammatory disease [104••]. The “low grade” inflammation is believed to be induced by obesity, metabolic syndrome, innate immunity, and aging-related inflammation, supporting emerging arguments for the inflammatory theory of OA [104••].
These concepts reveal the need for a more radical, comprehensive, and biology-based approach to studying OA, as has been emphasized by Qvist et al. [103], and the need for new analytical tools, reagents, and technology to help us study the metabolic processes and molecular changes underlying the structural alterations of OA [105, 106].
Diagnosis of OA is usually on the basis of clinical and radiography changes, which occur during quite late-stage disease and have poor sensitivity for monitoring disease progression [107]. This means OA is diagnosed during its irreversible stages, when treatment can be expected only to reduce pain and slow progression. This is a major disadvantage for drug development. The focus of DMOAD development must shift toward validation of biochemical and imaging biomarkers [108]: biomarkers might provide relevant information more rapidly than imaging techniques (i.e. radiography and MRI) [107]. Finally, emerging evidence suggests that OA is a disease of the whole joint, affecting many of its musculoskeletal components, including articular cartilage, tendons, muscles, ligaments, subchondral bone, and even adipose tissue [3, 31, 32•, 109]. DMOAD development will need to consider the contribution of each of these joint components to the disease.
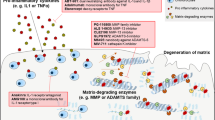
This paper has focused on better-targeted pharmacological therapy. The main targets discussed are summarized in Fig. 2.

Schematic diagram summarizing current concepts in pharmacological treatment of OA
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Goldring MB, Goldring SR. Osteoarthritis. J Cell Physiol. 2007;213(3):626–34.
Bay-Jensen AC, Slagboom E, Chen-An P, Alexandersen P, Qvist P, Christiansen C, et al. Role of hormones in cartilage and joint metabolism: understanding an unhealthy metabolic phenotype in osteoarthritis. Menopause. 2013;20(5):578–86.
Mahjoub M, Berenbaum F, Houard X. Why subchondral bone in osteoarthritis? The importance of the cartilage bone interface in osteoarthritis. Osteoporos Int. 2012;23 Suppl 8:841–6.
• Sellam J, Berenbaum F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat Rev Rheumatol. 2010;6(11):625–35. Important review that highlights the role of synovitis in the pathophysiology of OA.
Goldring MB, Berenbaum F. The regulation of chondrocyte function by proinflammatory mediators: prostaglandins and nitric oxide. Clin Orthop Relat Res. 2004;(427 Suppl):S37–46.
Berenbaum F, Eymard F, Houard X. Osteoarthritis, inflammation and obesity. Curr Opin Rheumatol. 2013;25(1):114–8.
Zhuo Q, Yang W, Chen J, Wang Y. Metabolic syndrome meets osteoarthritis. Nat Rev Rheumatol. 2012;8(12):729–37.
Stevens-Lapsley JE, Kohrt WM. Osteoarthritis in women: effects of estrogen, obesity and physical activity. Womens Health (Lond Engl). 2010;6(4):601–15.
• Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012;64(6):1697–707. This recent review highlights the fact that OA is a disease of the whole joint as an organ.
Loeser RF. Aging and osteoarthritis. Curr Opin Rheumatol. 2011;23(5):492–6.
• Loeser RF. Aging processes and the development of osteoarthritis. Curr Opin Rheumatol. 2013;25(1):108–13. This very recent review discusses the role of aging in the development of OA.
Loeser RF. Aging and osteoarthritis: the role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthr Cartil. 2009;17(8):971–9.
•• Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. This article outlines the senescence-associated secretory phenotype of tumor cells.
Felson DT. Developments in the clinical understanding of osteoarthritis. Arthritis Res Ther. 2009;11(1):203.
Flood J. The role of acetaminophen in the treatment of osteoarthritis. Am J Manag Care. 2010;16(Suppl Management):S48–54.
Vane JR, Botting RM. Mechanism of action of aspirin-like drugs. Semin Arthritis Rheum. 1997;26(6 Suppl 1):2–10.
Lipsky PE. Role of cyclooxygenase-1 and -2 in health and disease. Am J Orthop (Belle Mead NJ). 1999;28(3 Suppl):8–12.
Griffin MR. Epidemiology of nonsteroidal anti-inflammatory drug-associated gastrointestinal injury. Am J Med. 1998;104(3A):23S–9S. discussion 41S–2S.
Furst DE. Meloxicam: selective COX-2 inhibition in clinical practice. Semin Arthritis Rheum. 1997;26(6 Suppl 1):21–7.
Hochberg MC. COX-2 selective inhibitors in the treatment of arthritis: a rheumatologist perspective. Curr Top Med Chem. 2005;5(5):443–8.
Dooley M, Spencer CM, Dunn CJ. Aceclofenac: a reappraisal of its use in the management of pain and rheumatic disease. Drugs. 2001;61(9):1351–78.
Loewen PS. Review of the selective COX-2 inhibitors celecoxib and rofecoxib: focus on clinical aspects. CJEM. 2002;4(4):268–75.
Zhang W, Moskowitz RW, Nuki G, Abramson S, Altman RD, Arden N, et al. OARSI recommendations for the management of hip and knee osteoarthritis, part I: critical appraisal of existing treatment guidelines and systematic review of current research evidence. Osteoarthr Cartil. 2007;15(9):981–1000.
Suri S, Walsh DA. Osteochondral alterations in osteoarthritis. Bone. 2012;51(2):204–11.
Findlay DM. Vascular pathology and osteoarthritis. Rheumatology (Oxford). 2007;46(12):1763–8.
Castaneda S, Roman-Blas JA, Largo R, Herrero-Beaumont G. Subchondral bone as a key target for osteoarthritis treatment. Biochem Pharmacol. 2012;83(3):315–23.
• Goldring MB, Goldring SR. Articular cartilage and subchondral bone in the pathogenesis of osteoarthritis. Ann N Y Acad Sci. 2010;1192:230–7. This review article focuses on the specific articular cartilage and skeletal features of OA and the putative mechanisms involved in their pathogenesis.
Burr DB. Anatomy and physiology of the mineralized tissues: role in the pathogenesis of osteoarthrosis. Osteoarthr Cartil. 2004;12(Suppl A):S20–30.
Hunter DJ, Hellio Le Graverand-Gastineau MP. How close are we to having structure-modifying drugs available? Med Clin N Am. 2009;93(1):223–34. xiii.
Hunter DJ, Hellio Le Graverand-Gastineau MP. How close are we to having structure-modifying drugs available? Rheum Dis Clin N Am. 2008;34(3):789–802.
Abramson SB, Attur M. Developments in the scientific understanding of osteoarthritis. Arthritis Res Ther. 2009;11(3):227.
• Lories RJ, Luyten FP. The bone-cartilage unit in osteoarthritis. Nat Rev Rheumatol. 2011;7(1):43–9. This paper focuses on the functional unit formed by articular cartilage and subchondral bone in the context of OA.
Walsh DA, Chapman V. Bisphosphonates for osteoarthritis. Arthritis Res Ther. 2011;13(5):128.
Rosen CJ. Pathogenesis of osteoporosis. Baillieres Best Pract Res Clin Endocrinol Metab. 2000;14(2):181–93.
Burr DB, Gallant MA. Bone remodelling in osteoarthritis. Nat Rev Rheumatol. 2012;8(11):665–73.
Tanamas SK, Wluka AE, Pelletier JP, Martel-Pelletier J, Abram F, Wang Y, et al. The association between subchondral bone cysts and tibial cartilage volume and risk of joint replacement in people with knee osteoarthritis: a longitudinal study. Arthritis Res Ther. 2010;12(2):R58.
Findlay DM. If good things come from above, do bad things come from below? Arthritis Res Ther. 2010;12(3):119.
Rizzoli R. Osteoporosis: non-hormonal treatment. Climacteric. 2007;10 Suppl 2:74–8.
Spector TD. Bisphosphonates: potential therapeutic agents for disease modification in osteoarthritis. Aging Clin Exp Res. 2003;15(5):413–8.
Spector TD, Conaghan PG, Buckland-Wright JC, Garnero P, Cline GA, Beary JF, et al. Effect of risedronate on joint structure and symptoms of knee osteoarthritis: results of the BRISK randomized, controlled trial [ISRCTN01928173]. Arthritis Res Ther. 2005;7(3):R625–33.
Bingham 3rd CO, Buckland-Wright JC, Garnero P, Cohen SB, Dougados M, Adami S, et al. Risedronate decreases biochemical markers of cartilage degradation but does not decrease symptoms or slow radiographic progression in patients with medial compartment osteoarthritis of the knee: results of the two-year multinational knee osteoarthritis structural arthritis study. Arthritis Rheum. 2006;54(11):3494–507.
Felson DT, Kim YJ. The futility of current approaches to chondroprotection. Arthritis Rheum. 2007;56(5):1378–83.
Rizzoli R, Reginster JY, Boonen S, Breart G, Diez-Perez A, Felsenberg D, et al. Adverse reactions and drug-drug interactions in the management of women with postmenopausal osteoporosis. Calcif Tissue Int. 2011;89(2):91–104.
O’Donnell S, Cranney A, Wells GA, Adachi JD, Reginster JY. Strontium ranelate for preventing and treating postmenopausal osteoporosis. Cochrane Database Syst Rev. 2006;4, CD005326.
• Reginster JY, Badurski J, Bellamy N, Bensen W, Chapurlat R, Chevalier X, et al. Efficacy and safety of strontium ranelate in the treatment of knee osteoarthritis: results of a double-blind, randomised placebo-controlled trial. Ann Rheum Dis. 2013;72(2):179–86. This study describes the results of a double-blind, randomised placebo-controlled clinical trial on the safety and efficacy of strontium ranelate for the treatment of knee OA.
Richette P, Roux C. Impact of treatments for osteoporosis on cartilage biomarkers in humans. Osteoporos Int. 2012;23 Suppl 8:877–80.
Sutton S, Clutterbuck A, Harris P, Gent T, Freeman S, Foster N, et al. The contribution of the synovium, synovial derived inflammatory cytokines and neuropeptides to the pathogenesis of osteoarthritis. Vet J. 2009;179(1):10–24.
Clutterbuck AL, Asplin KE, Harris P, Allaway D, Mobasheri A. Targeting matrix metalloproteinases in inflammatory conditions. Curr Drug Targets. 2009;10(12):1245–54.
Cuzzocrea S. Role of nitric oxide and reactive oxygen species in arthritis. Curr Pharm Des. 2006;12(27):3551–70.
Abramson SB. Nitric oxide in inflammation and pain associated with osteoarthritis. Arthritis Res Ther. 2008;10 Suppl 2:S2.
Miyasaka N, Hirata Y. Nitric oxide and inflammatory arthritides. Life Sci. 1997;61(21):2073–81.
de Isla NG, Mainard D, Muller S, Stoltz JF. In vitro effects of diacerein on NO production by chondrocytes in response to proinflammatory mediators. Biomed Mater Eng. 2008;18(1 Suppl):S99–S104.
Fidelix TS, Soares BG, Trevisani VF. Diacerein for osteoarthritis. Cochrane Database Syst Rev. 2006;1, CD005117.
Liu D, Li H, Lu J, Bai Y. Tissue-specific implications of mitochondrial alterations in aging. Front Biosci (Elite Ed). 2013;5:734–47.
Lopez-Armada MJ, Carames B, Martin MA, Cillero-Pastor B, Lires-Dean M, Fuentes-Boquete I, et al. Mitochondrial activity is modulated by TNFalpha and IL-1beta in normal human chondrocyte cells. Osteoarthr Cartil. 2006;14(10):1011–22.
Blanco FJ, Lopez-Armada MJ, Maneiro E. Mitochondrial dysfunction in osteoarthritis. Mitochondrion. 2004;4(5–6):715–28.
Safdar A, Hamadeh MJ, Kaczor JJ, Raha S, Debeer J, Tarnopolsky MA. Aberrant mitochondrial homeostasis in the skeletal muscle of sedentary older adults. PLoS One. 2010;5(5):e10778.
Johnson K, Jung A, Murphy A, Andreyev A, Dykens J, Terkeltaub R. Mitochondrial oxidative phosphorylation is a downstream regulator of nitric oxide effects on chondrocyte matrix synthesis and mineralization. Arthritis Rheum. 2000;43(7):1560–70.
• Blanco FJ, Rego I, Ruiz-Romero C. The role of mitochondria in osteoarthritis. Nat Rev Rheumatol. 2011;7(3):161–9. This is a thought-provoking article that discusses the emerging role of mitochondria in OA.
Maneiro E, Martin MA, de Andres MC, Lopez-Armada MJ, Fernandez-Sueiro JL, del Hoyo P, et al. Mitochondrial respiratory activity is altered in osteoarthritic human articular chondrocytes. Arthritis Rheum. 2003;48(3):700–8.
Da Sylva TR, Connor A, Mburu Y, Keystone E, Wu GE. Somatic mutations in the mitochondria of rheumatoid arthritis synoviocytes. Arthritis Res Ther. 2005;7(4):R844–51.
Grishko VI, Ho R, Wilson GL, Pearsall AW 4th. Diminished mitochondrial DNA integrity and repair capacity in OA chondrocytes. Osteoarthr Cartil. 2009;17(1):107–13.
Kim J, Xu M, Xo R, Mates A, Wilson GL, Pearsall AW 4th. Mitochondrial DNA damage is involved in apoptosis caused by pro-inflammatory cytokines in human OA chondrocytes. Osteoarthr Cartil. 2010;18(3):424–32.
Johnson K, Svensson CI, Etten DV, Ghosh SS, Murphy AN, Powell HC, et al. Mediation of spontaneous knee osteoarthritis by progressive chondrocyte ATP depletion in Hartley guinea pigs. Arthritis Rheum. 2004;50(4):1216–25.
Afonso V, Champy R, Mitrovic D, Collin P, Lomri A. Reactive oxygen species and superoxide dismutases: role in joint diseases. Joint Bone Spine. 2007;74(4):324–9.
Meister A. Glutathione-ascorbic acid antioxidant system in animals. J Biol Chem. 1994;269(13):9397–400.
Meister A. Glutathione, ascorbate, and cellular protection. Cancer Res. 1994;54(7 Suppl):1969s–75s.
Scott JL, Gabrielides C, Davidson RK, Swingler TE, Clark IM, Wallis GA, et al. Superoxide dismutase downregulation in osteoarthritis progression and end-stage disease. Ann Rheum Dis. 2010;69(8):1502–10.
Ruiz-Romero C, Calamia V, Mateos J, Carreira V, Martinez-Gomariz M, Fernandez M, et al. Mitochondrial dysregulation of osteoarthritic human articular chondrocytes analyzed by proteomics: a decrease in mitochondrial superoxide dismutase points to a redox imbalance. Mol Cell Proteomics. 2009;8(1):172–89.
Mobasheri A. Role of chondrocyte death and hypocellularity in ageing human articular cartilage and the pathogenesis of osteoarthritis. Med Hypotheses. 2002;58(3):193–7.
Goggs R, Carter SD, Schulze-Tanzil G, Shakibaei M, Mobasheri A. Apoptosis and the loss of chondrocyte survival signals contribute to articular cartilage degradation in osteoarthritis. Vet J. 2003;166(2):140–58.
Vaamonde-Garcia C, Riveiro-Naveira RR, Valcarcel-Ares MN, Hermida-Carballo L, Blanco FJ, Lopez-Armada MJ. Mitochondrial dysfunction increases inflammatory responsiveness to cytokines in normal human chondrocytes. Arthritis Rheum. 2012;64(9):2927–36.
Cillero-Pastor B, Carames B, Lires-Dean M, Vaamonde-Garcia C, Blanco FJ, Lopez-Armada MJ. Mitochondrial dysfunction activates cyclooxygenase 2 expression in cultured normal human chondrocytes. Arthritis Rheum. 2008;58(8):2409–19.
Lotz M. Osteoarthritis year 2011 in review: biology. Osteoarthr Cartil. 2012;20(3):192–6.
Fernandez-Moreno M, Tamayo M, Soto-Hermida A, Mosquera A, Oreiro N, Fernandez-Lopez C, et al. mtDNA haplogroup J modulates telomere length and nitric oxide production. BMC Musculoskelet Disord. 2011;12:283.
• Morris BJ. Seven sirtuins for seven deadly diseases of aging. Free Radic Biol Med. 2013;56:133–71. This is a general review that discusses the role of sirtiuns in diseases associated with aging.
Dave M, Attur M, Palmer G, Al-Mussawir HE, Kennish L, Patel J, et al. The antioxidant resveratrol protects against chondrocyte apoptosis via effects on mitochondrial polarization and ATP production. Arthritis Rheum. 2008;58(9):2786–97.
Mobasheri A, Henrotin Y, Biesalski HK, Shakibaei M. Scientific evidence and rationale for the development of curcumin and resveratrol as nutraceutricals for joint health. Int J Mol Sci. 2012;13(4):4202–32.
Mobasheri A. Intersection of inflammation and herbal medicine in the treatment of osteoarthritis. Curr Rheumatol Rep. 2012;14(6):604–16.
Cawston TE, Wilson AJ. Understanding the role of tissue degrading enzymes and their inhibitors in development and disease. Best Pract Res Clin Rheumatol. 2006;20(5):983–1002.
Werb Z. ECM and cell surface proteolysis: regulating cellular ecology. Cell. 1997;91(4):439–42.
Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92(8):827–39.
Davidson R, Waters J, Kevorkian L, Darrah C, Cooper A, Donell S, et al. Expression profiling of metalloproteinases and their inhibitors in synovium and cartilage. Arthritis Res Ther. 2006;8(4):124.
Milner J, Rowan A, Cawston T, Young D. Metalloproteinase and inhibitor expression profiling of resorbing cartilage reveals pro-collagenase activation as a critical step for collagenolysis. Arthritis Res Ther. 2006;8(5):R142.
Caterson B, Flannery CR, Hughes C, Little CB. Mechanisms involved in cartilage proteoglycan catabolism. Matrix Biol. 2000;19:333–44.
Ohuchi E, Imai K, Fujii Y. Membrane type 1 matrix metalloproteinase digests interstitial collagens and other extracellular matrix macromolecules. J Biol Chem. 1997;72:2446–51.
Knauper V, Lopez-Otin C, Smith B, Knight G, Murphy G. Biochemical Characterization of Human Collagenase-3. J Biol Chem. 1996;271(3):1544–50.
Poole AR, Nelson F, Dahlberg L, Tchetina E, Kobayashi M, Yasuda T, et al. Proteolysis of the collagen fibril in osteoarthritis. Biochem Soc Symp. 2003;70:115–23.
Mapp PI, Walsh DA, Bowyer J, Maciewicz RA. Effects of a metalloproteinase inhibitor on osteochondral angiogenesis, chondropathy and pain behavior in a rat model of osteoarthritis. Osteoarthr Cartil. 2010;18(4):593–600.
Patel KR, Scott E, Brown VA, Gescher AJ, Steward WP, Brown K. Clinical trials of resveratrol. Ann N Y Acad Sci. 2011;1215:161–9.
Ernst E. Complementary or alternative therapies for osteoarthritis. Nat Clin Pract Rheumatol. 2006;2(2):74–80.
Ernst E. Musculoskeletal conditions and complementary/alternative medicine. Best Pract Res Clin Rheumatol. 2004;18(4):539–56.
Ernst E. Complementary medicine. Curr Opin Rheumatol. 2003;15(2):151–5.
Ernst E. Complementary and alternative medicine in rheumatology. Baillieres Best Pract Res Clin Rheumatol. 2000;14(4):731–49.
Setty AR, Sigal LH. Herbal medications commonly used in the practice of rheumatology: mechanisms of action, efficacy, and side effects. Semin Arthritis Rheum. 2005;34(6):773–84.
Akhtar N, Haqqi TM. Current nutraceuticals in the management of osteoarthritis: a review. Ther Adv Musculoskelet Dis. 2012;4(3):181–207.
Long L, Soeken K, Ernst E. Herbal medicines for the treatment of osteoarthritis: a systematic review. Rheumatology (Oxford). 2001;40(7):779–93.
Whitehouse MW, Butters DE. Combination anti-inflammatory therapy: synergism in rats of NSAIDs/corticosteroids with some herbal/animal products. Inflammopharmacology. 2003;11(4):453–64.
Shane Anderson A, Loeser RF. Why is osteoarthritis an age-related disease? Best Pract Res Clin Rheumatol. 2010;24(1):15–26.
Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69(3):89–95.
•• Kraus VB, Burnett B, Coindreau J, Cottrell S, Eyre D, Gendreau M, et al. Application of biomarkers in the development of drugs intended for the treatment of osteoarthritis. Osteoarthr Cartil. 2011;19(5):515–42. This excellent article by the OARSI FDA Osteoarthritis Biomarkers Working Group discusses how biomarkers can lead to an improved understanding of the molecular processes in OA and how they may facilitate the process of drug discovery.
Lesko LJ, Atkinson Jr AJ. Use of biomarkers and surrogate endpoints in drug development and regulatory decision making: criteria, validation, strategies. Annu Rev Pharmacol Toxicol. 2001;41:347–66.
Qvist P, Bay-Jensen AC, Christiansen C, Dam EB, Pastoureau P, Karsdal MA. The disease modifying osteoarthritis drug (DMOAD): Is it in the horizon? Pharmacol Res. 2008;58(1):1–7.
•• Berenbaum F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!). Osteoarthritis Cartilage. 2013;21(1):16–21. This elegant paper by Berenbaum highlights the fact that OA is an inflammatory disease.
Mobasheri A, Lewis R, Ferreira-Mendes A, Rufino A, Dart C, Barrett-Jolley R. Potassium channels in articular chondrocytes. Channels (Austin). 2012;6(6):416–25.
Mobasheri A. Osteoarthritis year 2012 in review: biomarkers. Osteoarthr Cartil. 2012;20(12):1451–64.
Rousseau JC, Delmas PD. Biological markers in osteoarthritis. Nat Clin Pract Rheumatol. 2007;3(6):346–56.
Krasnokutsky S, Samuels J, Abramson SB. Osteoarthritis in 2007. Bull NYU Hosp Jt Dis. 2007;65(3):222–8.
Edmonds S. Therapeutic targets for osteoarthritis. Maturitas. 2009;63(3):191–4.
Acknowledgments
The research leading to these results has received partial funding from the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 305815. The author is the coordinator of the D-BOARD Consortium funded by European Commission Framework 7 program (EU FP7; HEALTH.2012.2.4.5–2, project number 305815, Novel Diagnostics and Biomarkers for Early Identification of Chronic Inflammatory Joint Diseases). The author’s research has also been supported by grants from the Biotechnology and Biological Sciences Research Council (BBSRC; grants BBSRC/S/M/2006/13141 and BB/G018030/1), the Engineering and Physical Sciences Research Council (EPSRC), the National Centre for the Replacement, Refinement, and Reduction of Animals in Research (NC3Rs; grant number: Mobasheri. A. 28102007), and the Wellcome Trust (Grant no. CVRT VS 0901).
The funding bodies that support the author’s research did not affect any aspect of the research that has been reviewed in this article. The funders had no role in the design of the review, the collection and selection of papers, the interpretation of the papers, the writing of the manuscript, or the decision to submit the manuscript.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Ali Mobasheri has received industrial grant support from Mars and Waltham.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by the author.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Osteoarthritis
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Mobasheri, A. The Future of Osteoarthritis Therapeutics: Targeted Pharmacological Therapy. Curr Rheumatol Rep 15, 364 (2013). https://doi.org/10.1007/s11926-013-0364-9
Published:
DOI: https://doi.org/10.1007/s11926-013-0364-9