Article Text

Abstract
The inflammatory arthropathies that include rheumatoid arthritis, the seronegative spondyloarthropathies and systemic lupus erythematosus are characterised by marked alterations in the architecture and structural integrity of peri-articular bone; however, the pattern and natural history of the skeletal changes differs in these conditions. In part, this can be attributed to differences in the primary anatomical site of the inflammation, but also there is evidence that there are differences in the biological properties and products produced by inflammatory tissues. This review will focus on recent advances in the understanding of the cellular and molecular mechanisms that contribute to the differential pattern of articular bone remodelling in these prototypical inflammatory forms of arthritis.
- Arthritis
- Inflammation
- Treatment
- Rheumatoid Arthritis
- Spondyloarthritis
Statistics from Altmetric.com
Role of osteoclasts in RA bone erosions
Multiple lines of evidence have established that osteoclasts, the cells that are essential for physiological bone resorption, mediate the focal articular bone destruction in rheumatoid arthritis (RA) and related forms of inflammatory arthritis. This includes the demonstration of cells with osteoclast phenotype on the bone surfaces adjacent to resorption sites at the bone pannus interface,1 ,2 but in addition in animal models of inflammatory arthritis mice with an inability to form osteoclasts are protected from focal articular bone destruction.3–5 Formation of osteoclasts requires a source of myeloid lineage osteoclast precursors and a favourable environmental milieu that directs the precursors down the pathway of osteoclast differentiation and activation. Previous studies have shown that the synovium from RA and related forms of destructive inflammatory arthritis contains osteoclast precursors, and importantly that this tissue is a source of multiple potent osteoclast-inducing cytokines and growth factors, including receptor activator of nuclear factor κB ligand (RANKL), the master regulator of osteoclastogenesis without which there is an inability to form osteoclasts.6–13
Role of RANKL and cytokines in regulating osteoclastogenesis
Recent studies have shown that RANKL is produced by multiple cell types under physiological conditions, including osteocytes, hypertrophic chondrocytes and osteoblast lineage stromal cells, and that osteocyte-derived RANKL is a major driver of physiological bone remodelling.14 ,15 In RA synovium, synovial fibroblasts and T cells are the major source of RANKL, and inhibition of RANKL or inactivation of RANKL or its receptor using genetic approaches protects animals from the development of osteoclast-mediated bone erosions in models of inflammatory arthritis.7–12 Further evidence implicating the role of RANKL in articular bone loss comes from the observed efficacy of denosumab (an anti-RANKL antibody) in attenuating the development and progression of erosions in RA patients.16 ,17
The synovium in inflammatory arthritis is also a source of cytokines and growth factors that are anti-osteoclastogenic and direct myeloid lineage commitment down alternative pathways of cytodifferentition. Figure 1 includes a partial list of pro and anti-osteoclastogenic factors.6 ,18 The anti-osteoclastogenic factors include both type I and type II interferons that are products of T cells, and previous studies have defined the molecular mechanisms by which these factors inhibit pathways of osteoclast differentiation.19 ,20 Of interest, analysis of synovial tissues from patients with systemic lupus erythematosus reveals a strong interferon signature, and it has been suggested that this may account for the characteristic non-erosive features of lupus arthritis that conforms to the pattern of so-called Jaccoud's arthritis.21 Further evidence supporting this speculation is provided by studies of Mensah et al,22 who showed that overexpression of interferon-α in an animal model of RA resulted in attenuation of osteoclast-mediated articular erosions. T cells also modulate osteoclastogenesis via cell–cell interactions. For example, in recent studies, Zaiss and colleagues23 ,24 have shown that T regulatory cells (that are deficient in the RA synovium) suppress osteoclastogenesis via the expression of CTLA-4 in addition to production of the anti-osteoclastogenic cytokines IL-4 and IL-10.

Role of cytokines in determination of myeloid cell fate. Cytokines act on myeloid lineage precursor cells and exert differential effects on osteoclastogenesis and macrophage commitment. Many of the pro-osteoclastogenic cytokines act indirectly on stromal cells or osteoblast linage cells to upregulate receptor activator of nuclear factor κB ligand (RANKL), the potent osteoclast-inducing factor. CFU, colony-forming unit; GM–CSF, granulocyte macrophage colony-stimulating factor; IFN, interferon; IL, interleukin; M–CSF, macrophage colony-stimulating factor; TNF, tumour necrosis factor.
Bone formation and repair in inflammatory arthritis
Although great attention has focused on the focal osteoclast-mediated articular bone resorption in RA and the spondyloarthropathies, an additional distinguishing feature of these two forms of inflammatory arthritis is the differential patterns of bone formation. In RA, there is a virtual absence of bone repair and formation in the untreated state. In contrast, there is evidence of focal regions of enhanced bone formation at sites of articular and spinal inflammation in the spondyloarthropathies. Recent insights into the regulation of bone formation have come from the dissection of the role of the wingless (Wnt)-signalling pathway in controlling osteoblast differentiation and bone formation.25–27 This pathway is activated by binding of Wnt ligands to a cell surface receptor complex composed of the LRP5/6 and frizzled proteins. Binding results in translocation of the transcription factor β-catenin to the nucleus where it interacts with genes that result in increased osteoblast-mediated bone formation (figure 2). Of particular interest has been the essential role played by inhibitors of this pathway that include among others sclerostin and the family of dickkopf (DKK) proteins. Sclerostin and local Wnt signalling are required for the osteogenic response to mechanical loading and parathyroid hormone, and osteocyte-derived sclerostin has been shown to regulate local bone remodelling via modulation of RANKL expression.28–32

Regulation of the canonical wingless (Wnt)/β–catenin signalling pathway. Wnt ligands are a family of soluble mediators that activate the canonical Wnt signalling pathway by binding to a receptor complex consisting of LRP5/6 and frizzled. This results in activation of β-catenin that is translocated to the nucleus where it interacts with other transcription factors to induce osteoblast differentiation and enhance bone formation. Dickkopf-1 (DKK-1) and sclerostin inhibit this pathway by preventing activation of the receptor by the Wnt ligands.
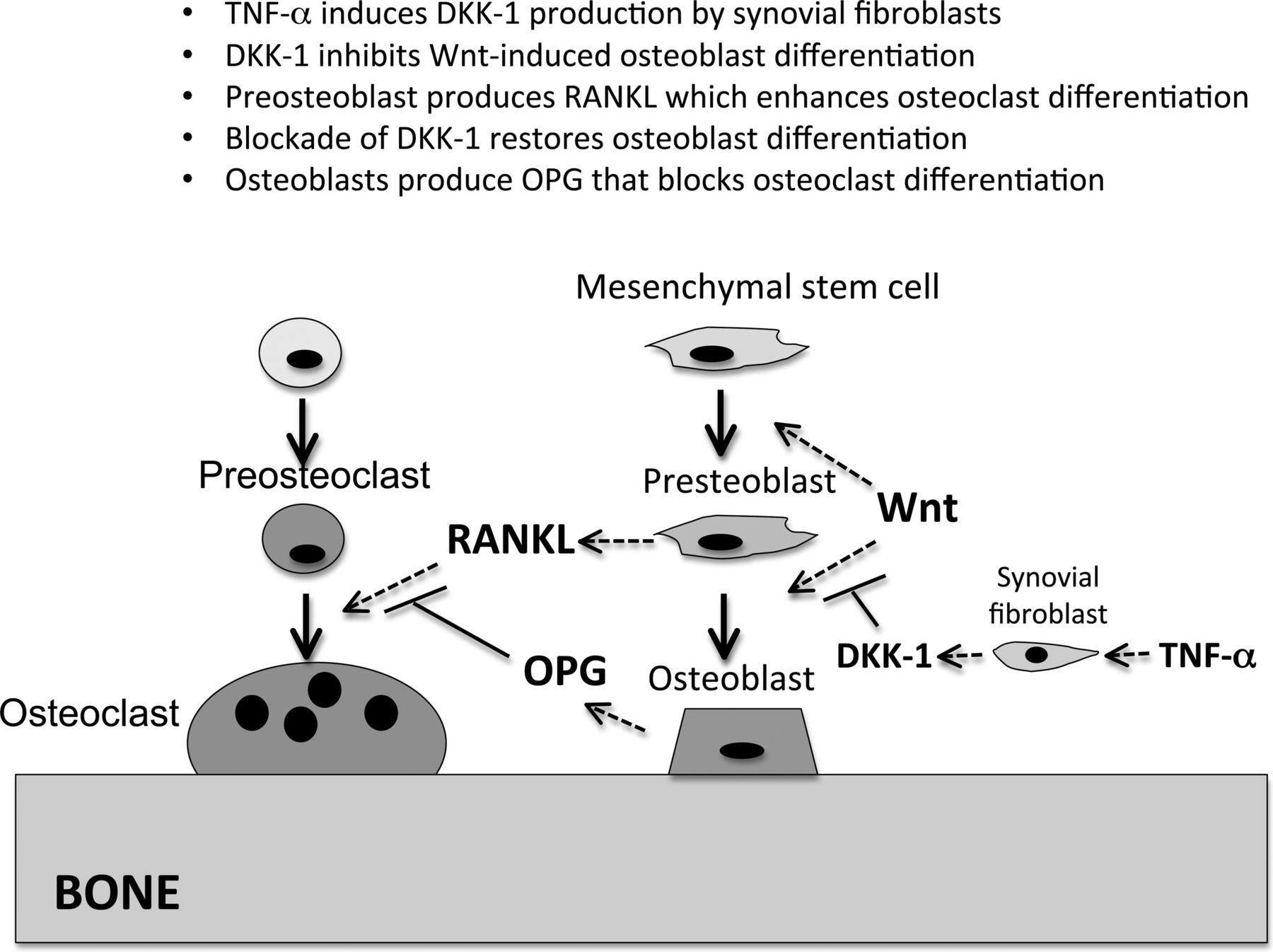
In a seminal paper by Diarra et al33 the authors showed that synovial tissues from patients with RA produced abundant levels of the potent Wnt pathway inhibitor DKK-1 and suggested that this could account for the defective bone repair. Using in-vitro cell culture, they showed that synovial fibroblasts were the principal source of the DKK-1 and that tumour necrosis factor α was a potent inducer of this inhibitor. They also showed that the local production of DKK-1 in the synovium was associated with elevations in serum levels, and that the serum DKK-1 levels correlated with disease activity. Of interest, patients treated with anti-tumour necrosis factor therapy showed a fall in DKK-1 levels. In contrast, they found that the serum levels of DKK-1 were not elevated in patients with ankylosing spondylitis, and they speculated that this could account for the tendency of those patients to develop excessive bone formation at sites of spinal and articular inflammation. Surprisingly, in animal models of RA they observed that inhibition of DKK-1 with a blocking antibody not only improved bone repair but also inhibited articular bone resorption, an effect they attributed to relief of the inhibitory constraint on osteoblast differentiation and upregulation of the production of osteoprotegerin, the potent inhibitor of RANKL33 ,34 (figure 3). More recently, Walsh et al35 have shown that RA synovium produces additional inhibitors of the Wnt pathway, including DKK-1, DKK-2, DKK-3 and soluble frizzled-related proteins 2 and 4. They showed that osteoblast function is impaired at sites of joint erosions in an animal model of RA and that there was evidence of the restoration of bone formation with treatment-induced resolution of inflammation.36
{kind=link}
{kind=link}
{kind=link}
Role of dickkopf (DKK)-1 in the regulation of bone remodelling in rheumatoid arthritis (RA) synovium. Tumour necrosis factor α (TNFα) produced by cells in the inflamed RA synovium induces DKK-1 expression in synovial fibroblasts leading to inhibition of wingless (Wnt)-dependent osteoblast differentiation resulting in suppression of bone formation and repair. Blockade of DKK-1 with an antibody relieves the constraints on activation of the Wnt/β–catenin pathway and restores osteoblast differentiation. β-catenin upregulates osteoprotegerin (OPG), resulting in inhibition of receptor activator of nuclear factor κB ligand (RANKL)-induced osteoclast-mediated bone resorption.
The effects of the Wnt ligands on osteoblasts and bone formation are mediated via the so-called canonical Wnt pathway that interacts with a receptor complex consisting of the LRP5/LRP6 and frizzle-related proteins25–27 (figure 2). Ligation of this receptor results in downstream signalling that leads to translocation of β-catenin to the nucleus where it enhances osteoblast differentiation and activity and upregulates osteoprotegerin (the inhibitor of RANKL) via transcriptional mechanisms. The canonical pathway is regulated by multiple Wnt family proteins including Wnt3a, which activates the pathway, and Wnt 5a that functions as an inhibitor. There is an additional non-canonical Wnt pathway that mediates effects via a single transmembrane receptor Ror1/2.37 With this receptor, the roles of the Wnt ligands are reversed, and Wnt3a functions as an inhibitor and Wnt5a as an agonist. In a recent study by Maeda et al38 they showed that synovial fibroblasts produce Wnt5a and that Wnt5a acts via Ror2 to upregulate RANK (the receptor for RANKL) on osteoclast precursors, resulting in enhanced osteoclastogenesis. Treatment with soluble Ror2 markedly attenuated osteoclast-mediated bone resorption in the collagen-induced model of RA, providing further evidence of the importance of the Wnt pathway in pathological bone remodelling and the potential utility of targeting components of this pathway to reduce the adverse effects of joint inflammation on local as well as systemic bone remodelling.
Role of the bone substrate in osteoclast differentiation
Of interest, although the RA synovium contains abundant osteoclast precursors that exist in an environmental milieu that is rich is osteoclastogenic cytokines and growth factors, cells with definitive features of osteoclasts are almost entirely restricted to the bone surface.1 ,2 This suggests that local factors in the immediate bone microenvironment, including components of the bone substrate, provide signals that are essential for terminal osteoclast differentiation and activation. The role of the substrate in osteoclast differentiation is supported by the studies of Saltel and coworkers39 who studied osteoclasts cultured on different surfaces and found that osteoclasts required a mineralised matrix to assume a fully differentiated phenotype.
In recent studies we have utilised an in-vitro osteoclast differentiation model to characterise the genes and gene products that are regulated by interaction of osteoclasts and their precursors with an authentic bone substrate.40 ,41 Expression profiling and hierarchical clustering was used to identify genes that were differentially upregulated in osteoclasts differentiated on the bone matrix compared to tissue culture plastic. Multiple genes were significantly changed by differentiation on the bone compared to plastic, and included among these were several genes that had previously been associated with osteoclasts, including for example the β3 integrin. We also identified clusters of genes previously not associated with osteoclasts. Among these genes, we focused our attention on annexin A8 (AnxA8) as a prototypical bone-regulated osteoclast gene, and showed by immunostaining that it was highly and selectively expressed in osteoclasts on bone surfaces associated with the RA pannus. AnxA8 is a member of a family of proteins that bind F-actin and phospholipids in a calcium-dependent manner42 ,43 and regulate cytoskeletal reorganisation, a process essential for osteoclast-mediated bone resorption. We subsequently used RNA interference to suppress AnxA8 expression and showed that this interfered with actin ring formation and resulted in loss of bone resorbing activity. These results provide evidence that in addition to soluble mediators and direct cell–cell interactions, the bone substrate also contributes to the regulation of osteoclast differentiation and activation, and importantly reveals additional targets for potential control of osteoclast-mediated bone resorption in inflammatory disorders.
References
Footnotes
-
Competing interest None.
-
Provenance and peer review Commissioned; externally peer reviewed.