Article Text

Abstract
Objectives: To characterise the catabolic response of osteoarthritic chondrocytes to Toll-like receptor (TLR) ligands.
Methods: Induction of the collagenases, matrix metalloproteinase (MMP)1 and MMP13, by TLR ligands was assessed in chondrocytes by real-time reverse transcriptase (RT)-PCR. TLR signalling pathway activation and their involvement in collagenase induction were confirmed by immunoblotting and use of pathway inhibitors and siRNA. TLR expression was compared in the femoral head cartilage of normal controls and patients with osteoarthritis (OA) by real-time RT-PCR.
Results: Ligands for TLR6/2 and TLR3 showed the greatest upregulation of MMP1 and MMP13 respectively, although all TLR ligands upregulated these MMPs. MMP1 and MMP13 induction by TLR3 and TLR1/2 or TLR6/2 ligands were dependent on Trif and MyD88, respectively. These inductions were dependent upon the nuclear factor (NF)κB pathway, but were differentially inhibited by various mitogen-activated protein kinase inhibitors, with MMP13 induction most reliant on the extracellular signal-regulated kinase pathway. In addition, ligands for TLR1/2 and TLR6/2, but not TLR3, induced significant collagenolysis in a cartilage resorption assay. Finally, TLR2 was significantly downregulated and TLR3 upregulated in OA, compared to normal, cartilage.
Conclusions: Activation of chondrocyte TLRs leads to differential collagenase gene activation. Treatment of chondrocytes with TLR1/2 or TLR6/2 ligands resulted in collagen resorption. The modulated expression of chondrocyte TLR2 and TLR3 in OA cartilage, compared to normal, may reflect a response to repair cartilage or prevent further extracellular matrix destruction. These data suggest modulation of TLR-mediated signalling as a potential therapeutic strategy for the treatment of OA.
Statistics from Altmetric.com
Rheumatoid arthritis (RA) and osteoarthritis (OA) are complex diseases that are still relatively poorly understood. Multiple genetic and/or environmental factors are thought to interact to trigger disease onset, and although progress has been made in the identification of genetic risk factors,1 2 the environmental factors are less well characterised. Despite significant improvements in RA treatments following the development of therapeutic anticytokine biologicals,3 these do not always prevent cartilage and bone destruction, the overriding debilitating factor.
Chondrocytes are the only cartilage cell type, precisely arranging the extracellular matrix (ECM) macromolecules to underpin normal tissue function and architecture,4 and are active players in destructive5 and reparative processes6 during disease. Pathological destruction results from homeostatic disruption via dysregulated proteolysis. Collagenases are unique enzymes that can all specifically cleave fibrillar collagens. They are matrix metalloproteinases (MMPs),7 and increases in collagenase-specific collagenolysis occur in pathological cartilage destruction8 resulting in essentially irreversible loss of tissue integrity.9 MMP activity is regulated at several levels including gene transcription, proenzyme activation, and active enzyme inhibition by tissue inhibitors of metalloproteinases (TIMPs).5 MMPs are induced by tumour necrosis factor α (TNFα) and interleukin 1 (IL1) and, in combination with the IL6-family cytokine oncostatin M (OSM), these proinflammatory cytokines synergistically enhance MMP production in vitro, ex vivo and in vivo.10–12
The recently discovered Toll-like receptor (TLR) family has increased awareness of the pivotal role innate immune signalling has in immunity to infection (reviewed in O’Neill and Brentano et al).13 14 Recognition of a diverse array of microbial ligands via TLRs and other innate recognition receptors is now known to be crucial in orchestrating appropriate effector mechanisms to combat pathogens.15 It is also clear these same recognition events can be triggered by endogenous ligands,16 resulting in inflammation and autoimmunity.17 Several major signalling pathways are TLR-activated including nuclear factor κB (NFκB) and mitogen-activated protein kinase (MAPK) pathways.13 Although signalling by each TLR has much in common, unique molecules and mechanisms provide specificity via different adaptor proteins (eg, MyD88, Mal, Trif and Tram) that activate signalling and mediate differential gene expression profiles.
Synovial injection of ligands for TLR2, 3, 4 or 9 induces or exacerbates arthritis in several experimental models.18–24 Furthermore, TLRs are expressed in synovium, and ligands detectable in inflamed synovium25 and recognised by TLR2 (eg, peptidoglycan) upregulate MMP1 expression in synovial fibroblasts.26 Although TLRs in synovium have been extensively studied, less is known about their function in cartilage. Different TLR profiles have been reported for chondrocytes, with only TLR2 consistently found.27–29 Chondrocyte TLR2 and 4 activation induces collagenases and subsequent resorption29 while TLR4-activation also increases IL1β mRNA and decreases aggrecan and type II collagen expression.28
We have shown that activation of several TLRs leads to collagenase expression in chondrocytes. Furthermore, ligands for TLR2-containing receptor complexes induced cartilage resorption and synergised with OSM. We conclude therefore, that the significantly reduced expression level of TLR2 in end-stage OA compared to normal cartilage, reflects a potential repair response to protect against further ECM destruction.
MATERIALS AND METHODS
Reagents
Recombinant human IL1α was a kind gift from GlaxoSmithKline (Stevenage, UK). Recombinant human OSM was from R&D Systems (Minneapolis, Minnesota, USA). TLR ligand Set I was from Apotech (Geneva, Switzerland) (table 1). Sulfasalazine, SP600125, SB203580 and U0126 were purchased from Calbiochem (Nottingham, UK). Primary antibodies used were phospho-extracellular signal-regulated kinase (ERK)1/2 (phospho-p44/42) (#9101), pan-ERK1/2 (p44/42) (#9102), phospho-p38 (#9211), phospho-c-Jun N-terminal kinase (JNK) (#9251) and NFκB inhibitor α (IκBα) (#9242) from Cell Signaling Technology (Danvers, Massachusetts, USA). A mouse monoclonal anti-glyceraldehyde 3′-phosphate dehydrogenase (GAPDH) antibody (clone 6C5; MAB374) was purchased from Chemicon (Chandlers Ford, Hampshire, UK). The polyclonal secondary immunoglobubins/HRP were from Cytomation (Dako, Glostrup, Denmark). Oligonucleotides and PCR Mastermix were purchased from Sigma-Genosys (Poole, Dorset, UK), while TaqMan primer/probes were from Applied Biosystems (Foster City, California, USA). Sybr green Mastermix was from Takara (Cambrex, Wokingham, UK). SmartPool siRNAs against Trif and MyD88 (see table 2), and siControl were from Dharmacon (Perbio, Cramlington, UK).
RNA extraction from cartilage samples and chondrocytes
Human articular cartilage was obtained from patients undergoing total joint replacement surgery. Hip cartilage samples from patients with OA were compared to cartilage from patients with a fracture to the neck of the femur (NOF) with no known history of joint disease essentially as described;30 NOF cartilages were lesion-free. Intact femoral heads were processed and RNA extracted using established methodology.31
Human articular chondrocytes (HACs) were isolated from OA knee joints by sequential digestion and cultured as described.32 When cells reached 80–90% confluence, the medium was removed and replaced with serum-free medium and the next day stimulated with test cytokines and/or TLR ligands at the indicated concentrations (table 1). Total RNA was isolated using a Cells-to-cDNA II kit (Ambion (Europe) Ltd., Huntington, UK). HACs were pretreated with selective pathway inhibitors for 30 min prior to stimulation. Total cell lysates were prepared using a modified Schindler buffer and Western blotted as previously described.33 This study was performed with Ethical Committee approval from Norfolk and Norwich University Hospital and Newcastle and North Tyneside Health Authority, and all patients provided informed consent.
Real-time reverse transcriptase (RT)-PCR
For TLR expression in cartilage, complementary DNA (cDNA) was synthesised from 0.5 μg of total RNA using Superscript II reverse transcriptase and random hexamers according to the manufacturer’s instructions (Invitrogen). Oligonucleotide primers and fluorescence-labelled probes are listed in table 2. TaqMan PCR conditions were as described previously.34 TLR1/2/4/6 (assay identification given in table 2) expression was determined with a TaqMan Low Density Array (Applied Biosystems) and normalised to the level of 18S rRNA gene expression using the calculation 2−ΔCT, where ΔCT represents CT(target gene) – CT(18S). For standard TaqMan/Sybr Green PCR, mRNA levels for each gene were obtained from standard curves, corrected using 18S rRNA levels. Cycling conditions with Sybr Green were 95°C 10 s, then 40 cycles of 95°C 5 s, 60°C 30 s and a standard dissociation curve analysis.
Gene silencing
Trif, MyD88 or siControl (non-depleting) siRNAs (100 nM) were transfected into HACs using Dharmafect 1 transfection reagent (Dharmacon) following the manufacturer’s protocol. Briefly, 1×104 cells were seeded into each well of a 96-well plate and transfected with the siRNA the next day. Cells were washed 24 h post transfection with PBS and cultured in serum-free medium for another 24 h prior to stimulation (24 h) with ligands. Total RNA was isolated and reverse-transcribed as above. Gene depletion was confirmed by real-time RT-PCR.
Immunoblotting
Human articular chondrocytes were grown to 80–90% confluence, serum-starved overnight, and then stimulated with cytokines for the indicated time periods. Cells were lysed with ice-cold buffer (50 mM Tris-HCl, pH 7.5; 1.2 M glycerol; 1 mM ethylene glycol tetraacetic acid (EGTA); 1 mM EDTA; 1 mM Na3VO4; 10 mM 2-glycerophosphate; 50 mM NaF; 5 mM sodium pyrophosphate; 1% (v/v) Triton X-100; 1 μM microcystin-LR; 0.1% (v/v) 2-mercaptoethanol; Roche protease inhibitor complex), particulate matter removed by centrifugation at 13 000 g, 5 min at 4°C, and lysates stored at –80°C until use. Lysates were resolved by sodium dodecyl sulfate polyacrylamide electrophoresis (SDS-PAGE), transferred to nitrocellulose membranes and subsequently probed using the antibodies described above. Immunoblot quantitation used the ImageQuant 1D software (GE Healthcare, Amersham, UK)
Cartilage assays
Dissected bovine nasal cartilage was cultured in serum-free medium (Dulbecco modified Eagle medium (DMEM) containing 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 2 mM glutamine, 100 μg/ml streptomycin, 100 IU/ml penicillin, 2.5 μg/ml gentamicin and 40 U/ml nystatin) overnight as described.11 Fresh medium, with or without treatments, was added (day 0). At day 7, culture supernatants were harvested and medium replaced. Cartilage and culture supernatants were again harvested at day 14 and residual cartilage papain digested. Hydroxyproline release was assayed as a measure of collagen degradation,35 and glycosaminoglycan release assayed as a measure of proteoglycan degradation.36 The extent of release was calculated as a percentage of the total. Collagenase activity in culture supernatants was determined by a 3H-acetylated collagen diffuse fibril assay with aminophenylmercuric acetate (0.67 mM) to activate procollagenases.37 Gelatinolytic activity was assayed by zymography.38
Statistical analysis
Significant differences between patient groups were determined using a two-sided Mann–Whitney U test. Standard TaqMan experiments were performed in at least triplicate for a minimum of three separate samples, with data analysed using a two-tailed Student t test. Cartilage experiments were performed in quadruplicate for three different cartilages, and significance assessed using analysis of variance (ANOVA) with post hoc Bonferroni multiple comparison test using commercial software (SPSS, V 11.0, SPSS Inc, Chicago, Illinois, USA).
RESULTS
TLR2 and TLR3 are differentially expressed between normal and osteoarthritic cartilage
An initial screen for TLRs (fig 1) found TLR2 expression in OA cartilage in line with previous reports,27–29 39 but significantly this expression was reduced (p<0.005) compared to normal (NOF). Conversely, TLR3 expression was significantly increased (p<0.005) in OA cartilage while TLR1, TLR4 and TLR6 expression levels were essentially unchanged. Generally, TLR2 expression in normal cartilage chondrocytes (NOF) was significantly higher than any other TLR analysed, with relative expression levels being: TLR2>>TLR4⩾TLR6>TLR1>TLR3 (fig 1). A real-time RT-PCR screen of isolated chondrocytes confirmed the expression of most TLRs, although TLR4 expression was very low and using this methodology TLR8 and TLR9 were undetectable (data not shown). Of those detected, TLR1 and TLR2 were upregulated by the procatabolic stimulus of IL1+OSM, while TLR7 was significantly downregulated (data not shown). As a control, we could detect TLR gene expression in peripheral blood cells (not shown).

TLR ligands selectively and differentially upregulate collagenase expression in chondrocytes
Collagenase expression induced by IL1 or TNFα is thought to be NFκB-dependent,40 and since TLRs also utilise NFκB, we determined whether TLR activation in HACs could stimulate similar expression. Most TLR ligands differentially induced MMP1 expression (fig 2A); the TLR6/2 ligand (L) (MALP-2) was the most consistent and robust stimulus. MMP13 was also induced by all ligands with the exception of the TLR9L (CpG) (fig 2A). The most robust stimulus for MMP13 was the TLR3L, poly(I:C). These mRNA data were reproducible at the protein level as determined by ELISA (data not shown). It is important to note the HAC data presented throughout are from different patients (individual figures are from a single patient/experiment) and although variations in terms of fold inductions by the various TLR ligands between chondrocyte populations (n = 10) were evident as previously reported,6 the overall effects were reproducible. Interestingly, responses for MMP1 or MMP13 induction by TLR6/2L and TLR3L, respectively were greater than for IL1+OSM, the most potent cytokine combination known to promote human cartilage degradation.11 Based on these observations, we selected the TLR1/2, TLR3 and TLR6/2 ligands for further study, and demonstrated dose-dependent induction of MMP1 and MMP13 gene expression. Most significantly, MMP13 induction via TLR3 activation even at low ligand concentrations exceeded that of IL1, while the TLR6/2L concentration could also be reduced without significantly altering the level of MMP1 induction (fig 2B).

TLR ligands synergise with OSM to induce collagenase expression
Similar to the ability of OSM to synergistically induce MMP1 and MMP13 expression in HACs when combined with IL1 or TNFα,6 10 41 we found that TLR1/2L and TLR3L also synergised with OSM; MMP1 and MMP13 levels were significantly higher than IL1+OSM although no significant synergy was observed with the TLR6/2L (fig 2C).
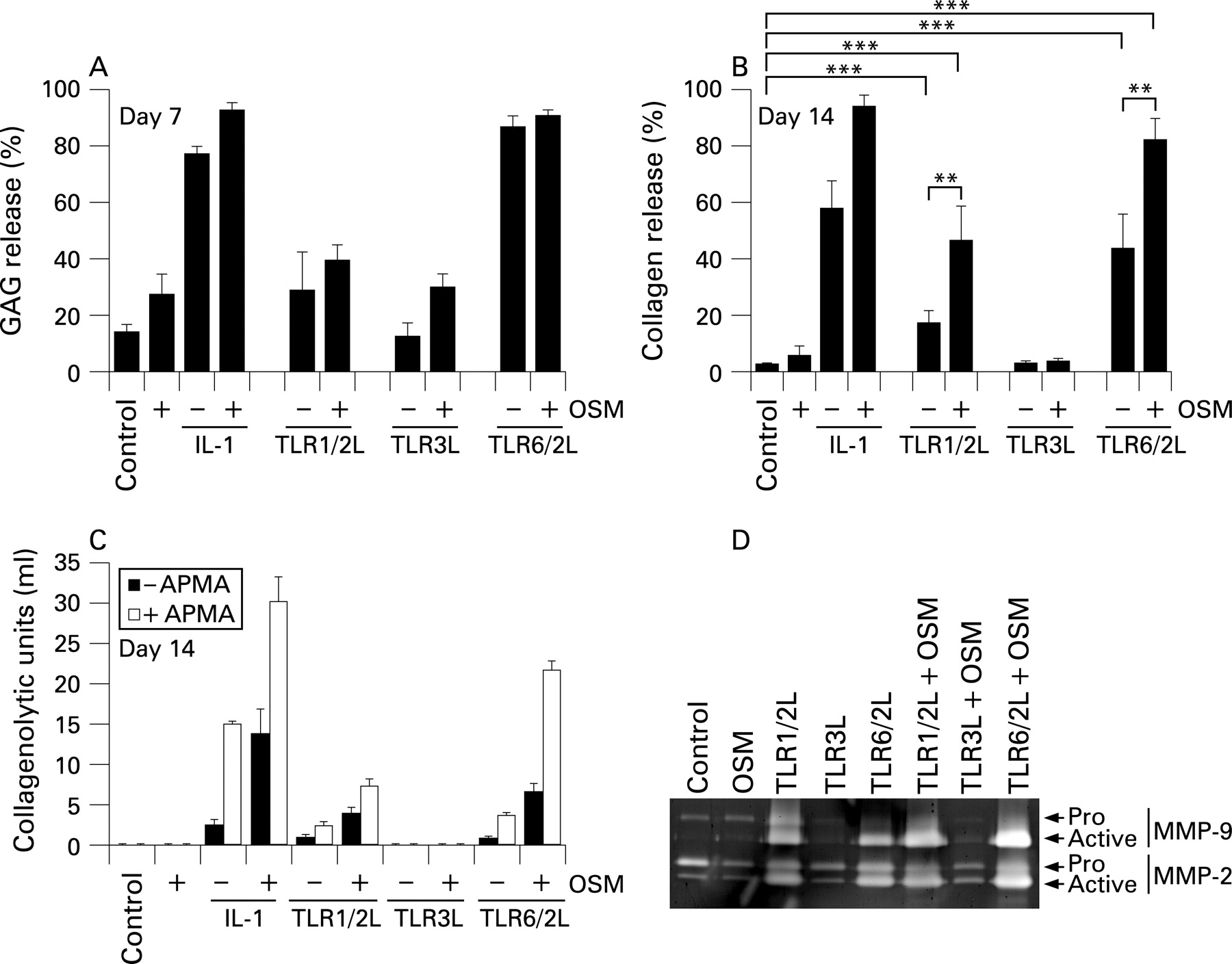
TLR1/2 and TLR 6/2 ligands promote cartilage resorption
TLR2 activation has been shown to induce cartilage resorption.29 We next examined the ability of ligands that act through TLR2 or TLR3 to resorb cartilage in an established model.11 Proteoglycan (GAG) release comparable to IL1+OSM was seen for the TLR6/2 ligand alone by day 7 and the TLR1/2 ligand by day 14 (fig 3A and not shown). IL1+OSM stimulation resulted in synergistic, and almost complete, collagenolysis as expected.11 Alone, TLR1/2 and TLR6/2 ligands promoted modest collagenolysis, while inclusion of OSM promoted synergistic release (fig 3B). The TLR3 ligand failed to promote any aggrecanolysis or collagenolysis, even with OSM (fig 3A,B). The observed TLR1/2 and TLR6/2 ligand-mediated collagenolysis correlated with total and active collagenolytic activity levels, most notably when acting synergistically with OSM (fig 3C). Similarly, inclusion of OSM to the TLR1/2 and TLR6/2 ligands enhanced detectable levels of active MMP9 and pro and active MMP2 in culture supernatants (fig 3D). The TLR3 ligand failed to modulate gelatinolytic or collagenolytic activities (fig 3C,D).
Different signalling pathways control TLR-induced MMP1 and MMP13 expression
TLR1/2, TLR3 and TLR6/2 ligands activated the NFκB pathway, as determined by IκBα degradation, and all three MAPK pathways (fig 4) but not the phosphatidylinositol 3-kinase pathway (not shown). However, the activation kinetics differed: the TLR6/2 ligand displayed the most rapid activation with complete IκBα degradation and maximal JNK, p38 and ERK phosphorylation by 30 min; the TLR1/2 ligand showed a more intermediate activation similar to that for IL1 although the activation was more prolonged; the TLR3 ligand activation was less pronounced than that of the other ligands or IL1 with only modest degradation of IκBα and MAPK phosphorylation by 90 min (fig 4B). This delay in pathway activation by TLR3L may be due to the endosomal location of the receptor.

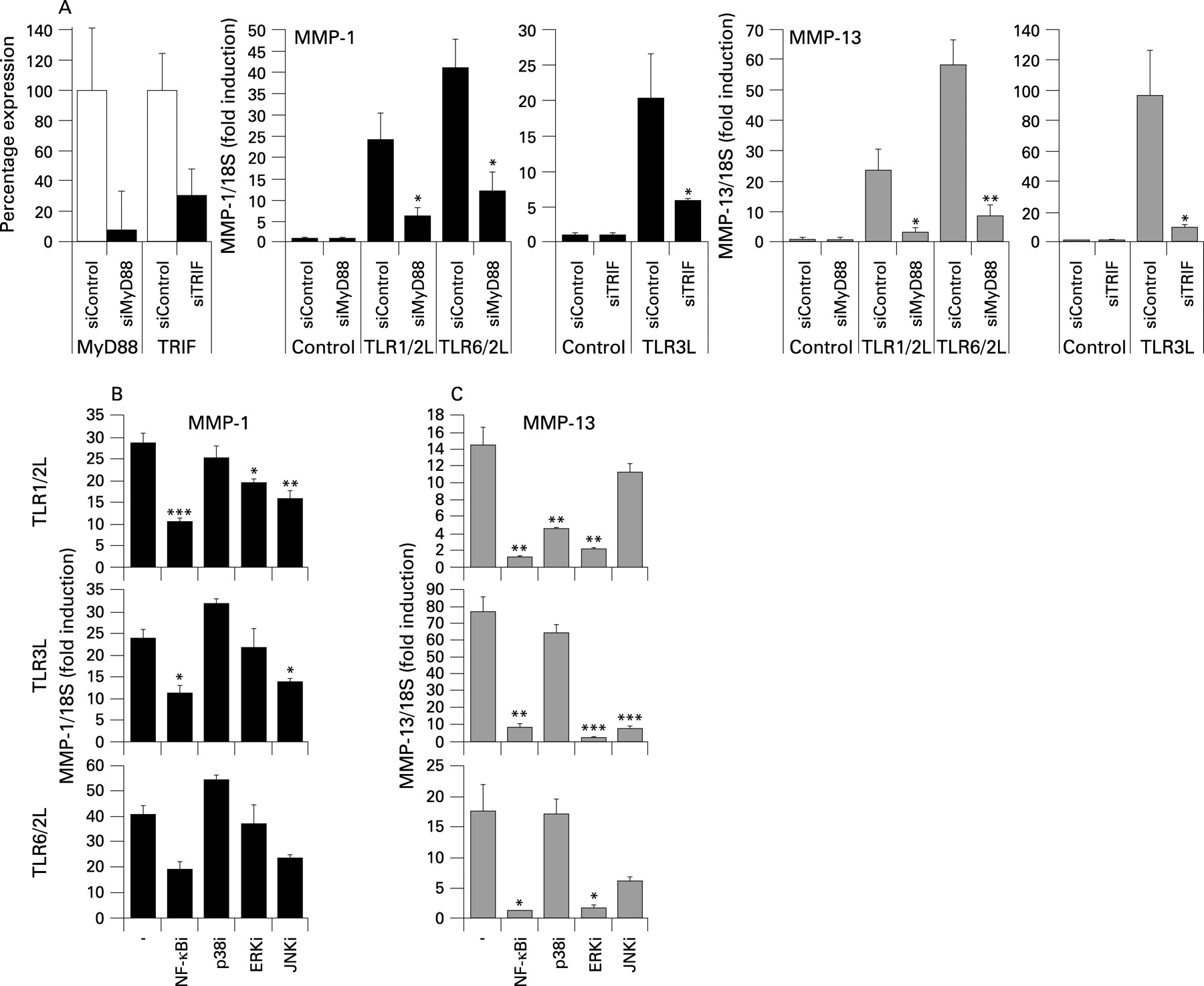
Gene silencing of MyD88 or Trif resulted in approximately ⩾70% depletion of their respective transcripts 48 h post transfection (fig 5A). As expected, MMP1 and MMP13 induction by the TLR1/2 and TLR6/2 ligands showed MyD88-dependence, while Trif depletion resulted in a significant decrease in MMP1 and MMP13 induction by the TLR3L (fig 5A).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The induction of MMP1 and MMP13 by the three ligands was sensitive to NFκB inhibition (fig 5B,C); notably, MMP13 induction appeared to be more NFκB-dependent than MMP1. Although the p38MAPK pathway was activated by all ligands, only MMP13 induction by the TLR1/2 ligand appeared p38-dependent (fig 5C). Conversely, ERK inhibition significantly reduced MMP13 expression by all the ligands but only modestly reduced TLR1/2L-induced MMP1 expression. Although MMP1 expression required JNK for maximal induction (fig 5B), only TLR3-induced MMP13 was strongly JNK-dependent (fig 5C).
DISCUSSION
A major finding of our study was the differential regulation of TLR2 and TLR3 in OA cartilage compared to normal. The marked downregulation of TLR2 in diseased cartilage may represent an attempt by chondrocytes to prevent further degradation since TLR2 activation, either alone29 or in combination with TLR1 or TLR6, promotes resorption. Our results show TLR3 activation increases MMP13 expression by chondrocytes, perhaps contributing to the increased MMP13 expression observed in OA cartilage (data not shown).34 We also confirmed the expression of TLR1, TLR4 and TLR6 genes although these were not differentially expressed between OA and normal cartilage. However, given that TLR1 and TLR6 associate with TLR2, the >10-fold reduction in TLR2 expression would significantly alter TLR1/2- and TLR6/2-stimulated effects in disease.
Since TLRs are innate immunity pattern recognition receptors, it is somewhat surprising that we, and others, have identified an extensive chondrocyte TLR repertoire.28 Kim et al showed TLR2 and TLR4 ligands, bacterial peptidoglycan and lipopolysaccharide (LPS), could induce MMP1, MMP3 and MMP13 in chondrocytes and increase aggrecan- and collagenolysis from cartilage.29 In our experiments, peptidoglycan (data not shown) and LPS induced MMP1 and MMP13 expression but not as robustly as TLR1/2, TLR3 or TLR6/2 ligands. We also failed to induce resorption with LPS although this may reflect differences in LPS preparations. TLR2 is reported to be upregulated in OA lesions,29 and this apparent discrepancy with our data probably reflects different methodologies (real-time RT-PCR vs immunohistochemistry) as well as regional gene expression differences; we screened RNA from “whole” articular cartilage containing normal and diseased areas. The expression pattern of TLR2 observed by Kim et al may actually represent a failure to downregulate TLR2 expression (as opposed to increased expression) leading to enhanced tissue destruction and hence lesions.29 TLRs can recognise endogenous ligands16 resulting from catabolism (see below), thus the downregulation of TLR2 in OA could be a self-limiting mechanism by which chondrocytes protect against subsequent catabolism, preserving ECM integrity for prolonged periods of time as is prevalent in OA.
Endogenous ligands recognised by TLR2 and TLR4 include heat shock protein 70 while TLR3 is activated by mRNA from necrotic cells.42 Monosodium urate or calcium pyrophosphate dehydrate microcrystals, found in synovium or articular cartilage associated with various arthropathies,27 43 signal through chondrocyte TLR2 to upregulate MMP expression and drive cartilage destruction.27 Recent data show that TLR4 inhibition reduces joint inflammation in the IL1 receptor antagonist knockout mouse model of spontaneous arthritis,44 suggesting TLR4 activation by endogenous arthritogenic ligands. A large group of potentially relevant endogenous ligands are ECM catabolites such as hyaluronan (HA) fragments,45 heparan sulfate,46 and fibronectin extra domain A.47 Known to be recognised by TLR2 or TLR4,16 these ligands are exposed during ECM injury/remodelling; in the lung, these TLRs drive inflammation in response to fragmented HA while protecting against inflammation-associated injury when HA is intact.45 Similarly, high molecular weight HA inhibits MMP expression and angiogenesis48 49 while IL1-induced hyaluronidase activity mediates HA fragmentation with concomitant loss of joint function;50 it will therefore be interesting to determine the effects of both HA forms on chondrocyte TLR activation. Recent findings indicate that the binding of exogenous ligands requires endogenous coligands, such as the dependence of Mrp8 and Mrp14 coligands for LPS activation of TLR4.51 Notably, Mrp8 and Mrp14, also called S100A8 and S100A9 respectively, are induced by chondrocytes in response to cytokine stimulation,6 are elevated in RA synovial fluid and may amplify proinflammatory cytokine responses within inflamed joints.52
Mouse gene deletion studies suggest reactive arthritis and streptococcal cell-wall fragment-induced joint inflammation are TLR4- and TLR2-dependent, respectively (both MyD88-dependent).18 53 Intra-articular injection of bacterial DNA (or CpG oligonucleotides) can induce a macrophage-mediated arthritis in mice,21 22 while systemic exposure to TLR ligands (in adjuvant) augments arthritis.18–20 Interestingly, peptidoglycans (recognised by TLR2) are present in RA synovium25 and upregulate MMP1 expression in synovial fibroblasts,26 strongly suggesting a role for TLRs in RA-associated joint degradation. TLR3 is trans-activated in response to necrotic synovial fluid cells54 and mice injected with dsRNA develop a self-liming arthritis.23 24 Our data confirm that TLR3 activation can markedly induce the expression of chondrocyte MMP13, yet the ligand used, poly(I:C), when added to cartilage, even in the presence of OSM, failed to promote resorption. This is likely due to a failure of this ligand to penetrate cartilage and/or ligand instability over the extended culture period since no induction of collagenase was detectable. However, TLR3 activation results in production of type I interferons (IFN),13 and poly(I:C) can suppress synovial proliferation and cytokine production via a type I IFN-dependent mechanism.55 This, coupled with our observation that IFNα blocks cytokine-induced cartilage resorption (data not shown), suggest that TLR3 ligands may prevent resorption implying that the increased TLR3 expression we observed in end-stage OA cartilage may be a “defence” mechanism to restrict further catabolism.
The differential regulation of MMP1 and MMP13 by different TLR ligands was marked, especially the significant TLR3-dependent induction of MMP13. Our data showed that MMP1 induction by the TLR ligands was less NFκB-dependent than for MMP13, substantiating a previous IL1 study.56 TLR3 signalling uses the adaptor Trif consistent with our data.57 Also, as expected, MyD88 depletion reduced the relative fold inductions of MMP1 and MMP13 by TLR1/2 and TLR6/2 ligands. MMP13 induction by the three selected ligands was critically dependent on ERK, with other MAPK pathways showing differential importance (induction by the TLR1/2L was p38 MAPK-dependent while the TLR3L required JNK). Together, these data confirm collagenase induction in chondrocytes depends on unique combinations of signalling pathways and crosstalk.40 Our study demonstrates that chondrocytes respond to a wide range of TLR ligands by inducing collagenase gene expression and cartilage destruction. Moreover, for the first time we have shown that TLR activation in the presence of OSM synergistically induces collagenase gene expression to levels that exceed those previously reported for the most potent inducer of human cartilage catabolism.11 Thus, specific TLR-induced collagenase expression can contribute to the cartilage catabolism prevalent in RA and OA, and modulation of chondrocyte TLR expression or activation may represent a mechanism to prevent cartilage destruction. Future work should aim to identify novel endogenous TLR ligands in synovial fluid of patients with OA and RA to determine the extent that TLR activation plays in disease progression.
REFERENCES
Footnotes
Funding: This work was funded by the Arthritis Research Campaign, Action Medical Research, the Dunhill Medical Trust and the JGW Patterson Foundation.
Competing interests: None declared.
Ethics approval: This study was performed with Ethical Committee approval from Norfolk and Norwich University Hospital and Newcastle and North Tyneside Health Authority, and all patients provided informed consent.