Article Text

Abstract
Objective To determine if withdrawing methotrexate (MTX) after 6 months of combination etanercept (ETN)+MTX, in MTX-inadequate responders with active rheumatoid arthritis (RA), is non-inferior to continuing ETN+MTX.
Methods Tumour necrosis factor-inhibitor naïve RA patients with disease activity score 28 (DAS28)≥3.2, swollen joint count≥3, despite stable MTX, were treated with ETN+MTX for 6 months, followed by randomisation to either continue ETN+MTX or switch to ETN monotherapy for an additional 18 months. The primary endpoint was change in DAS28 from 6-month randomisation to 12 months. The non-inferiority margin of change in DAS28 was 0.6, with prespecified analyses (DAS28<3.2 vs DAS28≥3.2).
Results 205 patients were randomised. DAS28 was stable in patients on ETN+MTX and increased slightly in patients on ETN monotherapy from 6 to 12 months. Non-inferiority was not achieved, with an adjusted difference of 0.4 (0.1 to 0.7) between the ETN and the ETN+MTX groups, for the month 6–12 change in DAS28. However, patients who achieved low disease activity (LDA; DAS28<3.2) at 6 months had a similar disease activity at 12 months, whether on monotherapy or combination therapy (DAS28 change 0.7 ETN vs 0.57 ETN+MTX, p=0.8148). Conversely, for patients who did not reach LDA at 6 months, those on ETN monotherapy had increased disease activity at 12 months, while disease activity continued to decrease for patients on combination therapy, at 12 months (DAS28 change 0.4 ETN vs −0.4 ETN+MTX, p=0.0023).
Conclusions Non-inferiority was not achieved. Withdrawing MTX after 6 months of continuation ETN+MTX in MTX inadequate responders did not yield the same degree of improvement between 6 and 12 months compared with continuing ETN+MTX.
Trial Registration ClinicalTrials.gov−NCT00654368.
- Rheumatoid Arthritis
- Methotrexate
- Anti-TNF
- DMARDs (biologic)
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Combination therapy with a biologic and methotrexate (MTX) usually yields better outcomes than biologic monotherapy in rheumatoid arthritis (RA).1–10 Despite this, in some patients, MTX is not well tolerated and patients frequently wish to discontinue it. A systematic literature review reported that rates of discontinuation for toxicity in those taking MTX ranged from 10% to 37%, with the most common adverse events (AEs) involving the gastrointestinal tract, liver, skin/hair, central nervous system, lungs, as well as cytopenias.11 Furthermore, a review of patient records from public and private drug plans in Quebec and Ontario, Canada, revealed that 54%–58% of patients prescribed a biologic with MTX did not take MTX.12 About 30% of patients are treated with biologic monotherapy as their first observed treatment,13 and data suggest that monotherapy with etanercept (ETN) is effective.14–16 Several other biologics are approved either in combination with disease-modifying antirheumatic drugs (DMARDs) such as MTX or as monotherapy.10
A previous study showed no significant difference at 16 weeks in disease activity score 28 (DAS28) improvement in MTX-inadequate responders (MTX-IRs) treated with ETN monotherapy versus ETN+MTX combination therapy (72.8% vs 75.2%, p=0.658) in a switch to ETN versus add ETN to MTX-IRs.16 ADORE16 suggests that efficacy of ETN monotherapy is comparable to ETN+MTX up to 4 months. However, the short duration to the endpoint may not have been sufficient to determine differences over time, and disease progression was not assessed radiographically.16
In contrast, another study2 ,3 reported better outcomes with ETN+MTX therapy. At 52 weeks, mean progression of erosion score (−0.2 vs 1.8; p=0.02) and American College of Rheumatology (ACR) 20%, 50% and 70% response rates were significantly better for ETN+MTX compared with ETN alone.2 However, probability of radiographic progression using probability plots was only marginally different between groups.
The objective of the Canadian Methotrexate and Etanercept Outcome (CAMEO) study was to determine whether ETN monotherapy is non-inferior to ETN+MTX by evaluating disease activity during an 18 -month period in MTX-IRs with active RA treated with an initial 6 -month course of ETN+MTX. Results up to 12 months (thus, 6 months after randomisation where participants either continued or discontinued MTX), which was the primary endpoint, are presented. Future analyses will determine the durability of response at 24 months, including proportion of dropouts and X-rays, and gene analyses that may be important for ongoing responsiveness.
Methods
Study design
This phase four, multicentre, open-label, unblinded randomised, 24-month trial was conducted at 27 centres across Canada in adults with RA who were MTX-IRs. Patients had active disease despite stable MTX therapy (≥15 mg/week (wk) or 10 mg/wk if intolerant) for >12 weeks and were tumour necrosis factor (TNF) inhibitor-naïve. Safety exclusion criteria included active infection and untreated latent tuberculosis. Patients were treated with ETN (50 mg/wk subcutaneously (SC))+MTX for 6 months, followed by randomisation to either ETN+MTX (at the same doses, with dose adjustments allowed for MTX) or ETN monotherapy (50 mg/wk SC) for an additional 18 months (figure 1A). ETN and MTX were obtained under usual standard of care (ie, no study drug was supplied by the sponsor). Canada has different rules by various drug plans and by each provincial government. In general, in order to obtain a TNFi, a patient must have failed high-dose MTX (≥20 mg/wk). Assessments occurred at screening, baseline, 6, 12, 18 and 24 months.

(A) Study design. (B) Patient disposition to month 12.
Institutional Review Board approval (Research Review Board, Inc., Richmond Hill, Ontario, Canada) and signed informed consent were obtained. The study was conducted in accordance with Canadian regulations and International Conference on Harmonisation (ICH) Good Clinical Practice (GCP) guidelines and registered at ClinicalTrials.gov registration #NCT00654368.
Patients
Patients met the following key inclusion criteria: age ≥18 years at baseline visit; ACR classification of RA, symptom onset of ≥6 months; active disease at baseline (≥3 swollen joints, DAS28≥3.2); no prior ETN therapy or any other TNF inhibitor; an indication for ETN as per approved product monograph; able to continue MTX and received a dose of ≥15 mg/wk (or 10 mg/wk if intolerant) for ≥12 weeks, with the dose being stable ≥4 weeks before the baseline visit; and ability to access ETN with private or public insurance. Key exclusion criteria included prior biologic treatment or any investigational therapy within 4 weeks of initiation of study medication or during the study period.
Endpoints
The primary endpoint was the between-group difference between ETN+MTX to ETN alone in change in DAS28 from 6-month randomisation to 12 months. Secondary endpoints for this portion of the trial included DAS28 improvement at 6 months; DAS28 remission (<2.6), low (<3.2) and high (≥3.2) scores at 6 and 12 months; Health Assessment Questionnaire Disability Index (HAQ-DI),17 Physician Global Assessment (PGA), Patient Global Assessment (PtGA) and pain Visual Analog Scale (VAS) (0=no pain; 100=very severe pain)18 at 6 and 12 months.
The patient-reported HAQ-DI assesses functional disability in daily activities across eight functional subscales: dressing, rising, eating, walking, hygiene, reaching, gripping and usual activities. Each question is scored according to four response options on a scale of 0–3: without any difficulty=0, with some difficulty=1, with much difficulty=2, unable to do=3, and the final score is the mean of the eight subscales.17 For PGA, the physician assessed patients’ activity of arthritis by marking a point on a 100-mm VAS that best described their condition (0=very good; 100=very poor). Similarly, patients assessed their own disease activity with a VAS in the PtGA.
Analysis subsets and covariates
The full analysis set (FAS) included all patients. The intent to treat (ITT) analysis included all randomised patients. The per protocol population is defined as all randomised patients with DAS28 measurements both at 6-month randomisation and 12 months. Baseline characteristics, disposition and reasons for study withdrawal were summarised descriptively for the FAS, ITT and per protocol populations. The primary analysis for non-inferiority was assessed based on the ‘per protocol’ population and a sensitivity analysis on the ITT population. Secondary outcomes were analysed on the FAS and ITT population.19
Statistical methods
Sample size was determined from a hypothesis of non-inferiority of ETN alone to ETN+MTX with respect to the mean DAS28 improvement from month 6 to month 12. The sample size of 90 patients randomised per arm was based on a one-sided 95% CI with 80% power, a non-inferiority margin of 0.6 change in DAS28 score and a common SD of 1.5 for a primary analysis on the per protocol population. Assuming 30% of enrolled patients would not reach randomisation (at 6 months), and a 10% attrition rate after month six, 250 patients were planned to be enrolled to ensure at least 180 patients randomised with 90 patients per arm.
The difference between the two treatment arms in the mean change in DAS28 from 6 to 12 months was calculated using the mean square error from analysis of variance adjusting for covariates at randomisation: private and public reimbursement; <2 and ≥2 years of disease duration; DAS28 <3.2 and ≥3.2. Secondary endpoints were summarised with descriptive statistics and two-sided 95% CIs. Proportion of patients achieving low disease activity (LDA) and remission as well as European League Against Rheumatism (EULAR) response at month 12 were summarised by relative risk (RR) (ETN+MTX relative to ETN) with 95% CIs. Missing data in the ITT analyses were assessed through sensitivity analyses using last observation carried forward (LOCF) non-responder imputation and multiple imputation. Results were similar for the various methods of handling missing data; LOCF results are presented here for simplicity.
Remission was defined as DAS28<2.6, LDA was defined as DAS28<3.2 and moderate-to-high disease activity (MHDA) was defined as DAS28>3.2.
Change in DAS28 was analysed on an ITT basis. Note that per protocol results for DAS28 are not shown but are consistent with ITT results.
Results
Patients
Two hundred and fifty-eight patients consented and started on ETN added to their stable MTX. At the time of initiation with ETN, DMARDs other than MTX were discontinued. Overall, 205 patients were randomised; 107 (41.4%) received ETN+MTX, 98 (38.0%) received ETN monotherapy and 53 (20.5%) were not randomised (figure 1B). Over the 6 months following randomisation, 21 (19.6%) patients in the combination group and 32 (32.7%) from the monotherapy group discontinued treatment. The most common reason for early withdrawal in the randomised groups was disease progression (ie, response achieved, then loss of response; or insufficient response achieved; or response achieved but disease flared). Eleven (10.3%) patients in the combination group and twenty (20.4%) in the monotherapy group withdrew due to disease progression. Four (3.7%) and six (6.1%) patients in the combination and monotherapy groups, respectively, withdrew due to AEs. The first patient was enrolled on 28 June 2008, and the study concluded in December 2012.
Disease characteristics and demographics at baseline and 6 months were stratified by the monotherapy and combination therapy treatment groups assigned at 6 months. Values were similar between the monotherapy and combination therapy groups at baseline and month 6, with the exception of mean HAQ score ((tables 1, 2 and see online supplementary table S1 (non-randomised population)).
Demographic and disease characteristics
Secondary outcome measures of disease activity (month 6 randomization and month 12)
Mean HAQ (95% CI) scores were numerically different at baseline (1.3 (1.1 to 1.3) vs 1.5 (1.4 to 1.6)) in the monotherapy group compared with the combination therapy group.
DAS28
Mean DAS28 for the ITT population at month 6 improved from 5.4 at baseline to 3.5 (figure 2A) for a mean change (95% CI) of −1.9 (−2.1 to −1.7).

(A) Change in disease activity score 28 (DAS28) from month 6 randomisation to month 12 (primary endpoint). (B) Change in DAS28 from month 6 randomisation to month 12—stratified by disease activity at 6 months.
From month 6 to 12, DAS28 was stable in patients on ETN+MTX and increased slightly in patients on ETN monotherapy. The change in DAS28 (95% CI) was 0.5 (0.3 to 0.7) for the ETN group and 0.04 (−0.2 to 0.3) for the ETN+MTX group (figure 2A). Non-inferiority was not achieved, with an adjusted difference of 0.4 (0.1 to 0.7) for the change in DAS28 between the ETN and the ETN+MTX groups (non-inferiority margin 0.6). However, an analysis in which patients were stratified by disease activity at 6 months showed that patients who achieved LDA at 6 months had a similar disease activity at 12 months whether on monotherapy or combination therapy. The change in DAS28 from 6 to 12 months was similar for both treatment groups whether on monotherapy or combination therapy (p=0.8148) (figure 2B). Conversely, for patients who did not reach LDA at 6 months, those on ETN monotherapy had slightly increased disease activity between 6 and 12 months, while disease activity continued to decrease for patients in the combination therapy group. For these patients, the change in DAS28 from 6 to 12 months was not similar between groups for ETN alone (0.4 (95% CI 0.1 to 0.7)) and ETN+MTX (−0.4 (−0.7 to −0.1); p=0.0023).
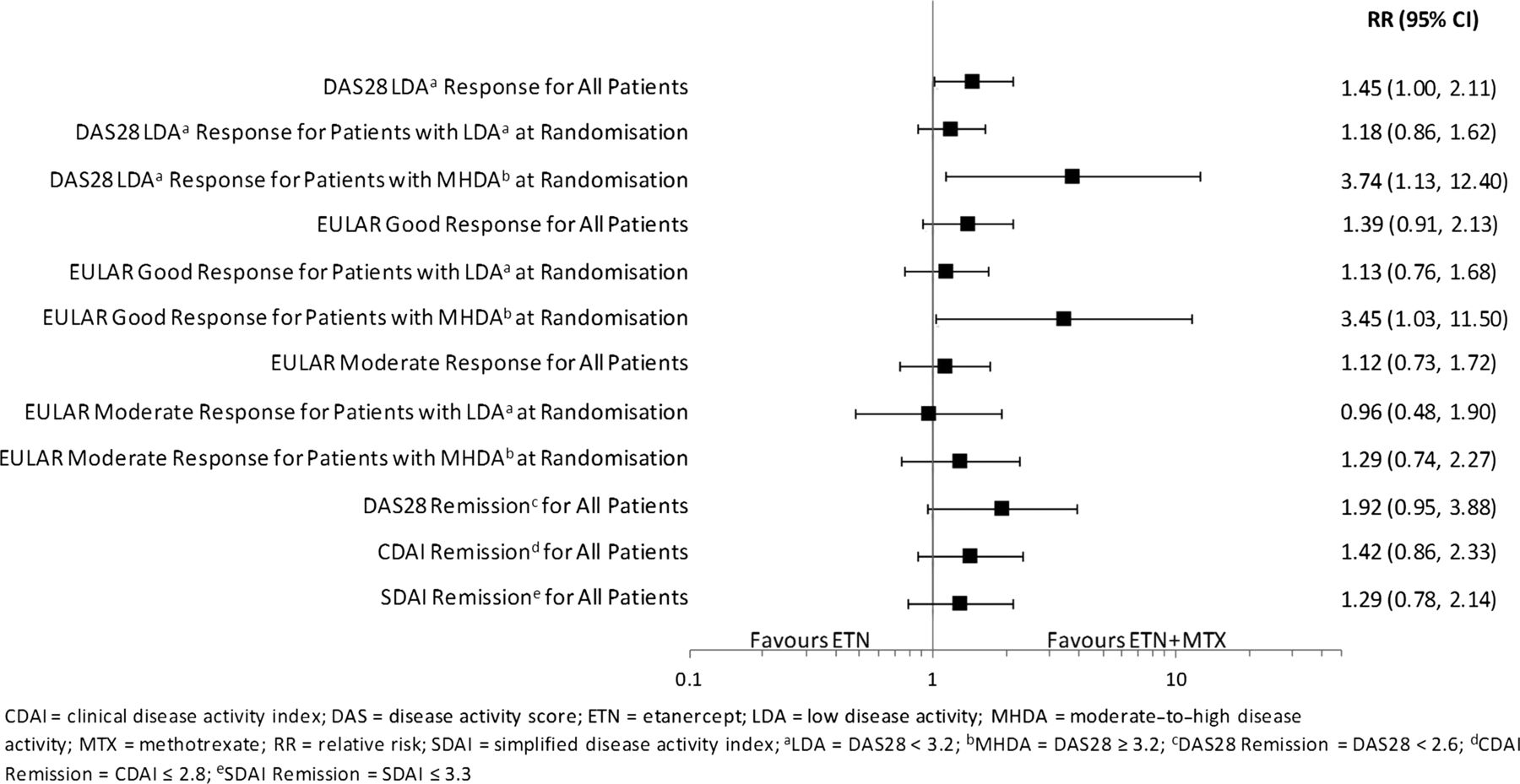
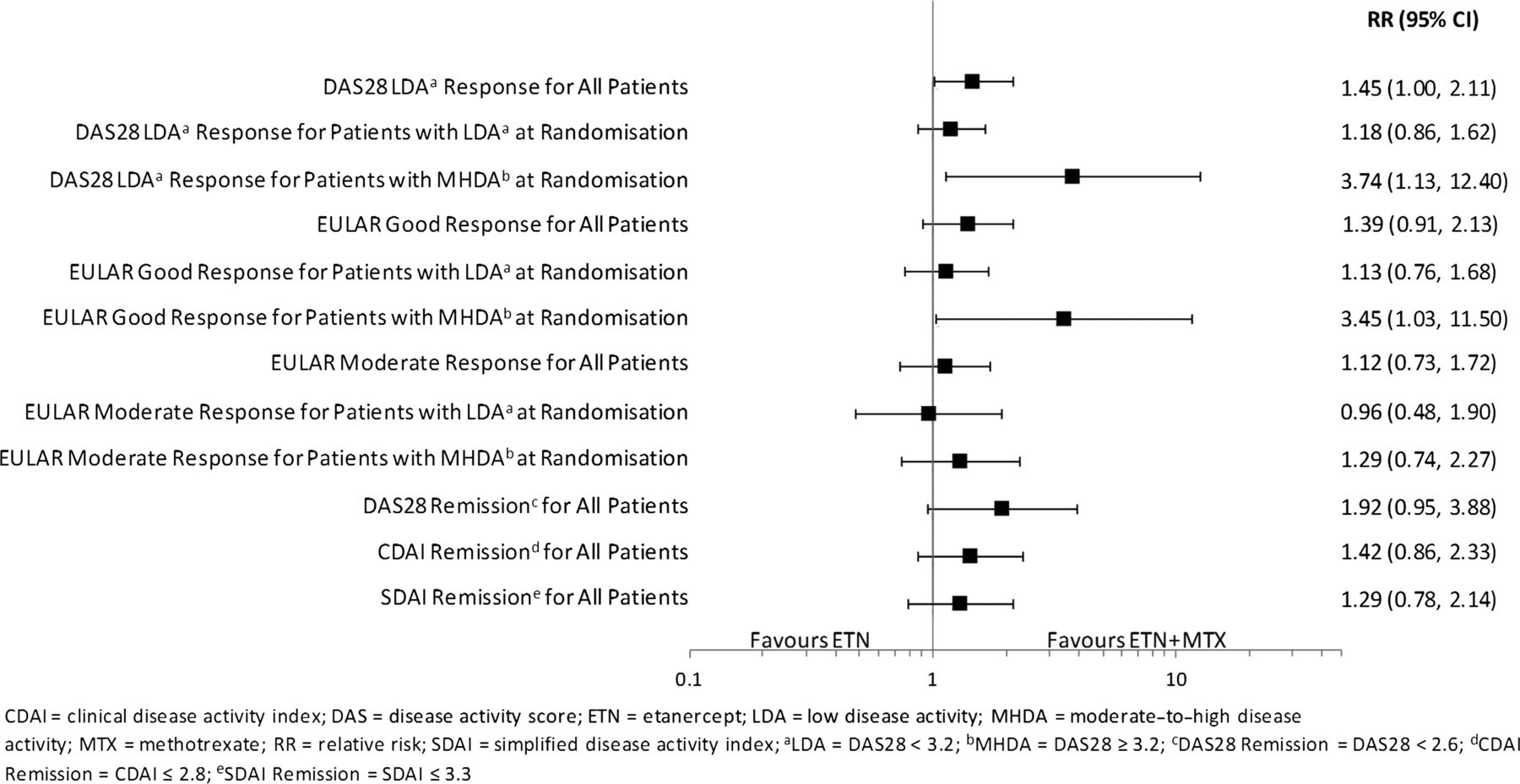
RRs (ETN+MTX relative to ETN) of randomised patients achieving LDA or remission at month 12 are shown in figure 3. At month 12, higher proportions of patients achieved LDA (RR (95% CI) 1.45 (1.00 to 2.11)]) and remission (RR (95% CI) 1.92 (0.95 to 3.88)) with combination therapy than monotherapy. In the subgroup of patients with LDA at 6 months, similar proportions achieved LDA or remission at 12 months in both treatment groups (LDA: RR (95% CI) 1.18 (0.86 to 1.62); REM: RR (95% CI) 1.38 (0.66 to 2.91)). However, for patients with MHDA at 6 months, the proportions were higher with combination therapy (LDA: RR (95% CI) 3.74 (1.13 to 12.40); REM: RR (95% CI) 6.03 (0.77 to 47.40)). Also see supplementary figure S1, web only content.
{kind=link}
{kind=link}
{kind=link}
Relative risk (ETN+MTX/ETN) of response from month 6 randomization to month 12.
EULAR response
RRs for EULAR response at 12 months for randomised patients are shown in figure 3. At month 12, proportions of patients with good or moderate EULAR responses were similar between the combination and monotherapy treatment groups, except for those with MHDA at 6 months. In this group of patients, a higher proportion achieved a good EULAR response at 12 months with combination therapy group compared with monotherapy (RR (95% CI) 3.45 (1.03 to 11.50)). Also see online supplementary figure S2.
HAQ, PtGA, PGA and pain VAS
At baseline, the mean HAQ-DI score (SD) was 1.4 (0.6) and it improved to 0.9 (0.7) at month 6. Thirty-eight per cent of patients had achieved a normal HAQ-DI score ≤0.5 at 6 months (see online supplementary figure S3a). The mean HAQ score (95% CI) was slightly better in the ETN group (0.8 (0.7 to 0.9)) than the ETN+MTX group (1.0 (0.9 to 1.1)) at month 6 (table 2). From months 6 to 12, HAQ scores worsened with monotherapy but did not change with combination therapy. At 12 months, mean HAQ score (1.0 (0.8 to 1.1) vs 1.0 (0.9 to 1.2)) and the proportion of patients with HAQ≤0.5 remained better for the monotherapy group compared with the combination therapy group (RR (95% CI) ETN+MTX/ETN=0.81 (0.55 to 1.20) (see online supplementary figure S3b).
At month 6, mean PtGA scores, PGA scores and pain VAS were similar between the ETN and the ETN+MTX groups (table 2). From months 6 to 12, mean PtGA, PGA and pain VAS scores worsened in the ETN group. PtGA and pain VAS scores remained stable in the ETN+MTX group, with some worsening in the PGA score. At 12 months, mean PtGA, PGA and pain VAS were similar between the ETN and the ETN+MTX groups.
Discussion
In this study, non-inferiority of ETN monotherapy compared with the ongoing combination of ETN and MTX, after 6 months of combination therapy in MTX-IRs, was not demonstrated. From 6 to 12 months, DAS28 was stable in patients on ETN+MTX and increased slightly in patients on ETN monotherapy. However, the mean difference between treatment groups was 0.45; thus, in some patients, the difference may not be clinically meaningful, whereas in others, their change in DAS28 after stopping MTX was clinically meaningful. One can potentially determine which patients may benefit from continuing MTX for the next 6 months vs stopping MTX. The subset of patients with LDA at 6 months achieved a similar DAS28 and EULAR response at 12 months whether on monotherapy or combination therapy. Conversely, patients with MHDA at 6 months performed better on combination therapy.
CAMEO, unlike other studies, focused on the disease state required to sustain ETN monotherapy regardless of the disease state at randomisation, with a prespecified outcome of patients achieving LDA. This is the first study that demonstrates the need to achieve a specific disease state in order to effectively discontinue MTX in MTX-IRs on combination therapy. Future data from this trial will evaluate the durability of monotherapy versus combination therapy and whether radiographic progression is different between the groups.
The data in this study are consistent with the JESMR study, where better clinical outcomes were observed with combination therapy, compared with monotherapy, even though the mean dose of MTX was very small. Rates of DAS28 remission and mean progression of erosion score were significantly better with ETN+MTX compared with ETN at 52 weeks in JESMR,2 though the probability of radiographic progression using probability plots was only slightly different between groups. In contrast, the ADORE study reported comparable efficacy between monotherapy and combination therapy16; however, the study had only a short follow-up period of 16 weeks. Other studies have examined tapering of treatment, following combination therapy. In an open-label extension study by Kremer et al,20 RA patients with MTX-IR treated with ETN+MTX were permitted dose reductions for MTX or, in some cases, corticosteroid after at least 3 months of combination therapy. Assessment at 3 years showed that 62% (n=41) of patients were able to discontinue or decrease the dose of MTX without apparent worsening of ACR response rates.20
The need for a low disease state or remission with combination of a biologic and MTX, before changes in therapy are attempted, is indirectly supported by studies of biologic dose reduction in MTX-IRs, such as the PRESERVE trial.21 Here, patients had to achieve a sustained period of LDA before dose reduction was attempted. For patients with moderate disease activity who then reached LDA/remission after 36 weeks of ETN+MTX, similar rates of LDA/remission were reported at 88 weeks, whether patients had continued on a full or half dose of ETN, combined with MTX.21 PRESERVE did not address whether patients would have done equally well on ETN monotherapy compared with combination therapy. Thus, CAMEO provides important information on the disease state required to stop MTX and maintain response with ETN monotherapy and suggests that a clinical response alone, such as change in DAS28 or an ACR response, is inadequate for such adjustment.
Some questions remain to be addressed, such as whether MTX in combination with ETN can yield longer-term durability compared with ETN monotherapy, and if so, what dose of MTX would be sufficient? Would sustainability improve if remission is achieved before MTX dose reduction? How long should remission be maintained for optimal results with dose reduction? Also, could MTX be reintroduced if effectiveness wanes, and could ETN also be reduced in those with long-term remission on combination or monotherapy with ETN?
The CAMEO study reflects real-world practice. Since it involved multiple centres, the drug was acquired in usual care, there were very few inclusion/exclusion criteria and many patients were on other background DMARDs at the time of ETN initiation where the non-MTX DMARDs were discontinued, the results are very generalisable. However, it has some limitations. The study was open label; the drop-out rate prior to randomisation was higher than expected, so more patients had to be enrolled to achieve an adequate number randomised. Although the per protocol population did not meet its target sample size at randomisation, ITT results are adequately powered and have consistent outcomes. CAMEO does not allow for comparison of outcomes based on length of time in remission, as the study lacks time points earlier than 6 months. However, the study continues for 18 months after randomisation, a total of 2 years, so more data of sustained remission will be obtained over time.
Gaining an understanding of long-term treatment is important in order to optimise disease control for patients with RA when using ETN therapy. Overall, maintaining MTX with ETN yields a better change in DAS28 than stopping MTX. However, for patients who achieve LDA by 6 months and desire to stop MTX, ETN monotherapy provides an effective alternative to combination therapy for the next 6 months.
Acknowledgments
We thank the following contributors, the clinical investigators of the CAMEO study, who provided and cared for study patients: Dr Algis Jovaisas, ON; Dr Ali Shickh, ON; Dr Andrew Chow, ON; Dr Anna Jaroszynska, ON; Dr Arthur Karasik, ON; Dr Boulos Haraoui, QC; Dr Carter Thorne, ON; Dr Eric Grant, NB; Dr Edward Keystone, ON; Dr Hyman Tannenbaum, QC; Dr Isabelle Fortin, QC; Dr Janet Markland, SK; Dr Janet Pope, ON (lead); Dr John Watterson, BC; Dr Juris Lazovskis, NS; Dr Louis Bessette, QC; Dr Majed Khraishi, NL; Dr Mark Hazeltine, QC; Dr Michel Gagné, QC; Dr Milton Baker, BC; Dr Murray Baron, QC; Dr Nabil Attie, QC; Dr Raman Joshi, ON; Dr Saeed Shaikh, ON; Dr Timothy McCarthy, MB; and Dr William Bensen, ON. We also thank Cheryl D'Abreo, MSc, Jessica Benzaquen, MSc, and Purnata Shirodkar, MSc, from The Synapse Group, Burlington, Ontario, who provided medical writing assistance as per authors’ directions and in accordance with standards set out by the International Committee of Medical Journal Editors. Writing support was funded by Amgen Canada and Pfizer Canada.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figures
- Data supplement 2 - Online table
Footnotes
-
Handling editor Tore K Kvien
-
Correction notice This article has been corrected since it was published Online First. Reference 17 has been deleted and subsequent references renumbered.
-
Contributors JEP acts as guarantor for this manuscript and accepts full responsibility for the work and/or the conduct of the study, had access to the data and controlled the decision to publish. JEP, BH, JCT and ECK contributed to the conception and design of the manuscript and the acquisition and interpretation of the data. AV contributed to the conception and design of the manuscript and the interpretation of the data. MP-C contributed to the conception and design of the manuscript and the analysis and interpretation of the data. All authors participated in drafting the manuscript as well as critically revising it throughout the writing process. Each author provided final approval of the current version of the manuscript that is to be published.
-
Funding This study was supported in part by Amgen Canada Inc. and Pfizer Canada. Amgen Canada Inc. oversaw the design, conduct and collection of data in the study and assisted in the analysis and interpretation of data.
-
Competing interests JEP has received grants and/or honoraria for her roles as investigator and for participating in advisory boards from Abbott Laboratories, Amgen Inc, Actelion, AstraZeneca Pharmaceuticals LP, Bristol-Meyers Squibb, Glaxo-Smith Kline, Hoffmann-LaRoche Inc, Janssen, Novartis Pharmaceuticals, Pfizer Pharmaceuticals, UCB. BH has received grants and/or honoraria for his roles as investigator, speaker and for participating in advisory boards from Abbott, Amgen, Bristol-Myers Squibb, Merck, Pfizer, Roche, UCB. JCT has received grants and/or honoraria for his roles as investigator and consultant from Amgen, Pfizer, Abbott, Bristol-Myers Squibb, Centocor Inc, Merck, Roche, UCB. AV and MP-C are employees of Amgen Canada Inc. and have stock options in Amgen Inc. ECK has received grants and/or honoraria for participating in advisory boards from Abbott Laboratories, Amgen Inc, AstraZeneca Pharmaceuticals LP, Bristol-Meyers Squibb, Centocor Inc, F. Hoffmann-LaRoche Inc, Genzyme, Merck, Novartis Pharmaceuticals, Pfizer Pharmaceuticals, UCB, Genentech Inc, Nycomed; and for his role as speaker from Abbott Laboratories, Bristol-Meyers Squibb, F. Hoffmann-LaRoche Inc, Merck, Pfizer Pharmaceuticals, UCB.
-
Ethics approval Research Review Board, Inc., 13085 Yonge Street, Unit 19, Suite 203, Richmond Hill, Ontario, Canada.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Prior Presentation Preliminary results of this study were presented at the following meetings: American College of Rheumatology Annual Scientific Meeting 2011 (Poster #1249), American College of Rheumatology Annual Scientific Meeting 2012 (Poster #1313), Paris Société Française de Rhumatologie Annual Meeting 2012 (Podium Presentation) and Canadian Rheumatology Association Annual Scientific Meeting 2013.