Article Text

Abstract
Objectives To report results of subgroup analyses of bone mineral density (BMD) and bone turnover markers from a randomised, double-blind, placebo-controlled, phase II study of denosumab, an investigational RANKL inhibitor, in patients with rheumatoid arthritis (RA) concurrently receiving treatment with bisphosphonates or glucocorticoids.
Methods Patients received subcutaneous placebo (n=75), denosumab 60 mg (n=71) or denosumab 180 mg (n=72) at baseline and 6 months. Assessments included dual x-ray absorptiometry scans of the lumbar spine and hip, and determination of levels of serum type I C-telopeptide (sCTx-I) and serum procollagen 1N-terminal peptide (P1NP).
Results Denosumab treatment increased mean lumbar spine and hip BMD and reduced sCTx-I and P1NP compared with placebo through 12 months, regardless of baseline BMD or marker levels or concomitant bisphosphonate or glucocorticoid use.
Conclusions This study extends evidence that denosumab increases BMD and reduces bone turnover in patients with RA and may provide a new therapeutic option for reducing systemic bone loss in patients with RA.
Statistics from Altmetric.com
Introduction
Inflammation in rheumatoid arthritis (RA) produces local and systemic effects including focal joint erosions, subchondral bone erosions and periarticular and systemic osteoporosis. These effects reflect an imbalance among the mediators of bone resorption and formation1 and increase the risk of fracture. Glucocorticoid use further increases fracture risk.2 These RA-specific risks combine with the known osteoporosis risk in older women, who represent a significant proportion of patients with RA.3 Bone resorption in RA is mediated by RANK ligand (RANKL),4 5 whose expression is critical for the development of structural damage in RA.6,–,8 Denosumab, an investigational, fully human monoclonal antibody, inhibits RANKL and thereby reduces bone resorption.9
In this phase II study, we evaluated denosumab in patients with RA who were receiving methotrexate. The effects of denosumab in reducing structural damage, bone mineral density (BMD), and bone turnover for the total population have been reported previously.10 This analysis describes changes in BMD and bone turnover markers in patients with RA treated with denosumab. To determine whether the effects of denosumab were influenced by other treatments that affect bone metabolism (glucocorticoids and bisphosphonates), we evaluated patients' changes in BMD and bone turnover markers according to concurrent glucocorticoid or bisphosphonate administration and compared them with results for patients not receiving those agents.
Methods
The study design and inclusion criteria have been reported previously10 and are described in the online supplementary data. Patients who had active RA for ≥24 weeks and were receiving methotrexate received denosumab 60 mg, denosumab 180 mg or placebo by subcutaneous injection at baseline and at 6 months. Glucocorticoids could be dosed at ≤15 mg/day of prednisone or equivalent at study entry, and could be added or dose-modified to a maximum of 15 mg/day of oral prednisone or equivalent, except ≤2 weeks before scheduled assessments. Bisphosphonates were allowed throughout the study. After the first 6 months, patients could receive rescue treatment for signs and symptoms of RA with any approved anti-tumour necrosis factor therapy. Randomisation was stratified by current use of glucocorticoids and prior use of biological agents.
Bone mineral density was assessed by dual x-ray absorptiometry scans of the lumbar spine and total hip at baseline, 1, 6 and 12 months. Fasting blood samples were obtained at baseline, 1, 3, 6 and 12 months, before administration of the study drugs. Serum type I C-telopeptide (sCTX-I) was assessed by Amgen using kits from Nordic Biosciences (Herlev, Denmark) (precision, 2–9% of the coefficient of variance (CV)). Safety parameters included adverse events, vital signs and laboratory values. Serum procollagen 1N-terminal peptide (P1NP) was assessed by a specialty laboratory (Covance, Chantilly, Virginia, USA) using kits from Orion Diagnostica (Espoo, Finland) (precision, 2–6% of the CV).
End points have been described previously.10 This report describes post hoc analyses: comparisons of changes in BMD and bone turnover markers from baseline through 12 months in patients who received systemic glucocorticoids compared with those who did not, and in patients taking oral bisphosphonates compared with those who were not. We also examined the correlation between BMD changes and baseline values of bone turnover markers. The glucocorticoid subgroup was defined as patients using ≥2.5 mg/day of prednisone (or equivalent) for ≥90 days during the study. The bisphosphonate subgroup included patients who received any bisphosphonate for any length of time during the study. Statistical methods are described in the online supplementary data.
Results
A total of 227 patients were enrolled; 218 received study treatment, and most (94%) completed treatment. Demographics and baseline characteristics were generally similar between treatment groups (table 1), except as noted later.
Demographic and baseline disease characteristics: concurrent glucocorticoid and bisphosphonate use and non-use
Glucocorticoid recipients included more men and were slightly younger than non-recipients. In this subgroup, the median prednisone dose used was 5 mg at baseline and 4 mg at the end of the study (range 1–15 mg at both time points). Patients receiving bisphosphonates were somewhat older than those not receiving bisphosphonates. Disease characteristics were similar in the glucocorticoid and bisphosphonate subgroups, with a mean RA duration of approximately 11 years; BMD T-scores were similar and within the normal range in both groups. Median baseline levels of the bone turnover markers sCTx-I and P1NP were lower among patients receiving concomitant bisphosphonates than in those not receiving bisphosphonates (sCTx-I, 0.2 ng/ml vs 0.40 ng/ml; P1NP, 26.4–35.6 µg/l vs 43.0–46.4 µg/l) (table 1).
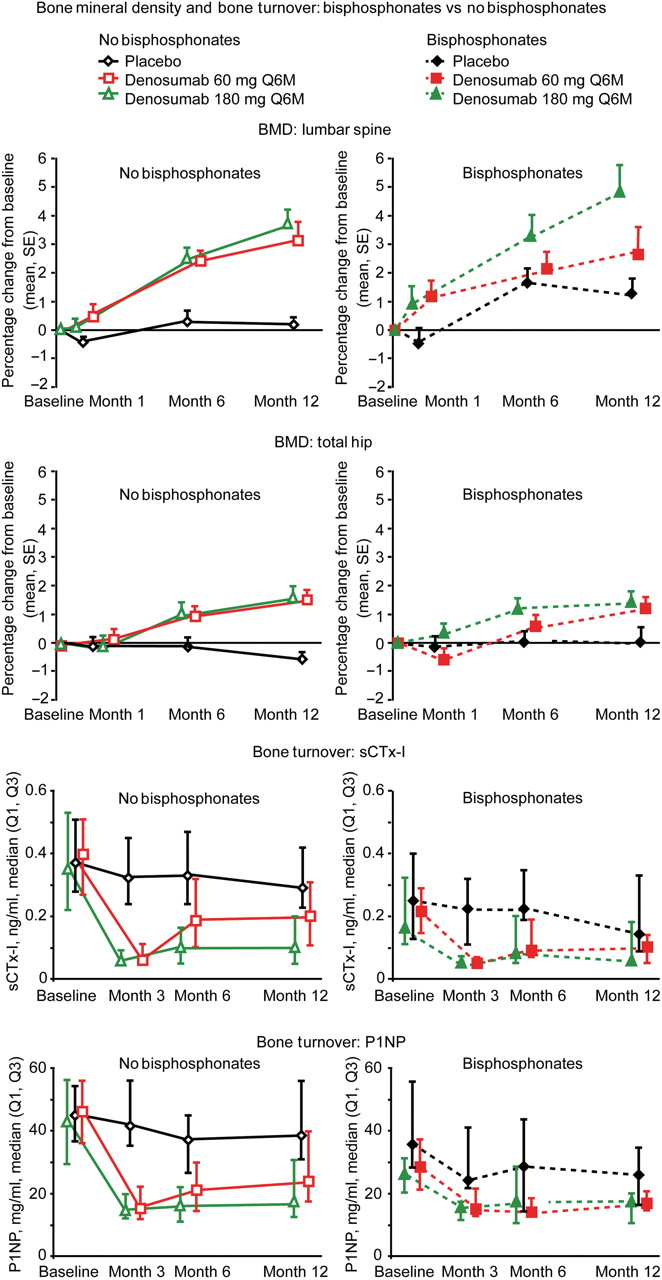
At 6 and 12 months, statistically significant increases in BMD compared with placebo (p<0.001) were seen at the lumbar spine and total hip in denosumab-treated patients (supplementary data figure S1). Similar denosumab-induced increases in BMD at the spine and total hip were seen regardless of glucocorticoid use (figure 1) or bisphosphonate use (figure 2).

Denosumab increased bone mineral density (BMD) at the lumbar spine and total hip and reduced levels of the bone turnover markers serum type I C-telopeptide (sCTx-I) and serum procollagen 1N-terminal peptide (P1NP), regardless of concomitant treatment with glucocorticoids. Denosumab or placebo was administered on study day 1 and at 6 months; BMD levels increased steadily from the first dose. Suppression of bone turnover markers was evident 3 months after the first dose; levels began to return to baseline levels as the end of each 6-month dosing interval approached. Q1, first quartile; Q3, third quartile.
{kind=link}
{kind=link}
Denosumab increased bone mineral density (BMD) at the lumbar spine and total hip and reduced levels of the bone turnover markers serum type I C-telopeptide (sCTx-I) and serum procollagen 1N-terminal peptide (P1NP), regardless of concomitant treatment with bisphosphonates. Denosumab or placebo was administered on study day 1 and at 6 months; BMD levels increased steadily starting with the first dose. Suppression of bone turnover markers was evident 3 months after the first dose; levels began to return to baseline levels as the end of each 6-month dosing interval approached. Q1, first quartile; Q3, third quartile.
Denosumab treatment also reduced sCTx-I and P1NP rapidly and consistently from baseline in all subgroups (figures 1 and 2). In denosumab-treated patients, reductions in sCTx-I and P1NP were evident at 3 months; levels increased as patients approached the end of each 6-month dosing interval. Although there was a trend toward a difference between denosumab and placebo treatment groups, statistical significance was not calculated because small subgroup sizes would limit the usefulness of these comparisons. Baseline P1NP, but not baseline sCTx-I, correlated with BMD increases over the course of treatment (for lumbar spine, regression correlation coefficient of baseline P1NP: 0.05, 95% CI 0.02 to 0.07, p<0.0001, for total hip; regression correlation coefficient of baseline P1NP: 0.02, 95% CI 0.00 to 0.04, p=0.03). During the study treatment phase, anti-tumour necrosis factor rescue treatment was administered to eight patients (4%), at similar rates in the denosumab and placebo groups and among glucocorticoid users and non-users.
Safety data from this study have been previously reported.10 Rates and types of adverse events were comparable between denosumab and placebo groups. The use of glucocorticoids or bisphosphonates did not affect the incidence or types of adverse events observed (data not shown). The incidence of clinical fractures during the 12-month treatment period was low (≤2%) and did not correlate with the use of denosumab, bisphosphonates or glucocorticoids. No cases of osteonecrosis of the jaw or clinically relevant changes in laboratory results, vital signs or parathyroid hormone levels were reported.
Discussion
In this study of patients with RA, denosumab increased BMD and reduced bone turnover compared with placebo in the overall population, in patients receiving concurrent glucocorticoids or bisphosphonates, and in patients not receiving those treatments.
Denosumab, like the bisphosphonates, is an antiresorptive agent used to treat osteoporosis, but its mechanism of action differs. Denosumab inhibits bone resorption by blocking interactions between RANK and its ligand, thereby preventing formation, activation and survival of osteoclasts. Bisphosphonates, in contrast, are incorporated into bone, inhibiting the ability of osteoclasts to resorb bone. The effects of this difference in mechanism of action were seen in two recent phase III studies of postmenopausal patients with low bone mass; in both studies, denosumab-treated patients experienced significantly greater BMD increases than patients treated with the bisphosphonate alendronate during the study, regardless of previous bisphosphonate treatment.11 12
Ninety patients (40%) received glucocorticoids during this study. Chronic glucocorticoid use is known to cause bone loss, which may be treated with bisphosphonates. In this study, glucocorticoid use (≤15 mg/day prednisone) did not appear to inhibit denosumab-induced increases in BMD; increases were similar for denosumab-treated patients in the glucocorticoid and non-glucocorticoid subgroups (figure 1).
In this study, patients for whom bisphosphonates had been previously prescribed could continue to receive bisphosphonate treatment regardless of their randomised, blinded treatment assignment; consequently, 29 patients received both denosumab and bisphosphonates. Although patients are unlikely to receive concomitant denosumab and bisphosphonates in clinical practice, evaluation of the concurrent use of these agents in patients with RA is important because the effects of bisphosphonates may persist after treatment discontinuation.13,–,15 In this subgroup, as expected, baseline values of sCTx-I among bisphosphonate-treated patients (about 0.2 ng/ml) were lower than those of other patient subgroups (0.3–0.4 ng/ml) (table 1), and were similar to those seen in the phase III study of women with osteoporosis previously treated with alendronate who were switched to denosumab.12 In both studies, patients who were treated with denosumab and a bisphosphonate had greater increases in BMD than patients treated with a bisphosphonate alone, even though their bone turnover rates were relatively low at baseline. No safety differences were observed in patients treated with denosumab or bisphosphonates compared with those not receiving bisphosphonates. Although the small subgroup sizes in this study (and the resulting potential for error) warrant caution in the interpretation of results, these data extend the evidence showing the potential utility of denosumab in patients previously treated with bisphosphonates.
Conclusion
Many patients with RA receive bisphosphonates, glucocorticoids, or both. This is the first study to characterise the effects of denosumab treatment in such patients, demonstrating that denosumab suppressed systemic bone loss beyond the suppression achieved with bisphosphonates and regardless of glucocorticoid use, with no unexpected patterns of adverse events overall or in any subgroup. In combination with previously reported reductions of joint erosions in this study, these results suggest that denosumab may provide a new therapeutic option for preventing loss of bone density in patients with RA.
Acknowledgments
In addition to the authors, we thank the following members of the Denosumab RA Study Group who served as investigators at the clinical sites and made important contributions to this study Christopher Atkins, Andre Beaulieu, Mary Bell, Stephen Bookbinder, Alan Brodsky, Atulya Deodhar, Gino Divittorio, Alan Fishman, Eric Grant, Brian Grimmett, Boulous Haraoui, Joseph Huffstutter, Eric Hurd, Alan Kaell, Arthur Kavanaugh, Jonathan Kay, Ed Keystone, Majed Khraishi, Michael Kohen, Mark Iannini, David Mandel, Robert McKendry, Phillip Mease, Larry Moreland, Sara Newell, Charles Peterfy, Jeffrey Poiley, Boris Ratiner, Christopher Ritchlin, Joy Schechtman, Michael Schiff, John Sharp, H Arthur Silverman, Gary Sultany, Hyman Tannenbaum, Robert Trapp, Peter Valen and Michel Zummer. We also thank Sue Hudson and Ting Chang for providing writing assistance on behalf of Amgen Inc.
References
Supplementary materials
Web Only Data ard.2009.112920
Files in this Data Supplement:
{kind=link}
Footnotes
-
Funding This study was funded by Amgen Inc. RKD has received research grants from Amgen, Merck, Eli Lilly and Roche. She has received consulting fees from Merck, Eli Lilly, Roche, UCB and Novartis and has been on the speakers' bureau for Merck, Eli Lilly, Roche, Procter & Gamble, Pfizer, Sanofi-Aventis and Novartis. SBC has received research grants and consulting fees from Amgen, Genentech, Biogen-IDEC, Merck, Sanofi-Aventis, Procter & Gamble, Pfizer, Centocor, Scios, Bristol Myers Squibb and Wyeth-Ayerst. NEL has received research grants and consulting fees from Proctor and Gamble, Merck and Co, Eli Lilly, Amgen, ZosanoPharma and Pfizer. WP has received research grants and/or consulting fees and/or served on speakers' bureaus for Pfizer, Genentech Inc, Bristol Myers Squibb, Wyeth, Roche, TAP, Abbott, Centocor and Amgen Inc.
-
Competing interests WS has served on speakers' bureaus for Amgen Inc, Centocor, Abbott, Bristol-Meyers Squibb and Genentech. LZ, HW, WT and RN are employees of Amgen and have received Amgen stock and stock options.
-
Ethics approval Approval was obtained from the institutional review boards and independent ethics committees of participating medical centres.
-
Provenance and peer review Not commissioned; externally peer reviewed.