Article Text

Abstract
Objectives Evaluate the efficacy of intravenous golimumab 2 mg/kg+methotrexate (MTX) in patients with active rheumatoid arthritis (RA) receiving MTX.
Methods Patients (n=592) with active disease (≥6/66 swollen, ≥6/68 tender joints, C-reactive protein ≥1.0 mg/dl, rheumatoid factor positive and/or anticyclic citrullinated protein antibody positive at screening) despite MTX (15–25 mg/week) participated in this double-blind, placebo-controlled, phase 3 study. Patients were randomised (2:1) to receive intravenous golimumab 2 mg/kg, or placebo infusions at weeks 0 and 4 and every (q) 8 weeks; patients continued MTX. Placebo patients with <10% improvement in combined swollen/tender joint counts at week 16 could early escape to intravenous golimumab 2 mg/kg. The primary endpoint was week 14 American College of Rheumatology 20% (ACR20) response. Analyses employed non-responder imputation and last-observation-carried-forward.
Results At week 14, significantly (p<0.001) larger proportions of golimumab+MTX than placebo+MTX patients achieved ACR20 response (59% vs 25%, respectively), a disease activity score of good/moderate (EULAR) response (81% vs 40%), and greater median improvement in health assessment questionnaire scores (0.500 vs 0.125). Improvements versus placebo+MTX were observed by week 2. Similar proportions of patients receiving golimumab+MTX and placebo+MTX, respectively, reported adverse events through week 16 (47% and 44%) and week 24 (53% and 49%). Serious adverse events were reported by more golimumab+MTX (4.1%) than placebo+MTX (2%) patients at week 24.
Conclusion The addition of intravenous golimumab rapidly and significantly improved signs and symptoms in patients with active RA despite ongoing MTX, in some patients by week 2.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Patients with rheumatoid arthritis (RA) and an inadequate response to disease-modifying antirheumatic drugs such as methotrexate (MTX) frequently have a tumour necrosis factor (TNF)-antagonist(s) added to the regimen.1 Golimumab, a newer human monoclonal antibody directed against TNF, has demonstrated safety and efficacy in large, randomised, double-blind, phase 3 trials in the treatment of MTX-naïve,2 MTX-experienced3 and anti-TNF-experienced4 patients with RA when administered monthly via subcutaneous injection.
Intravenous administration of golimumab every 12 weeks (q12 weeks) was previously assessed in a phase 3 trial in patients with RA, with persistent disease despite MTX therapy.5 While the study's primary endpoint (≥50% improvement in American College of Rheumatology (ACR50) response criteria at week 14) was not achieved (21.4% vs 13.2%, p=0.051), intravenous golimumab+MTX substantially reduced RA signs/symptoms and improved physical function, particularly beyond week 14. Moreover, intravenous golimumab 2 mg/kg at weeks 0 and 12, plus MTX, yielded a substantially higher ACR20 response at week 14 (55%, 71/129 patients) than placebo+MTX (28%, 36/129); no unexpected toxicities were observed.5 The current GO-FURTHER trial further assessed intravenous golimumab 2 mg/kg efficacy/safety in a larger patient population, while also evaluating a different dosing strategy (induction dosing at weeks 0 and 4, followed by q8-week maintenance therapy; see online supplementary file) to establish an efficacious dosing regimen for long-term maintenance therapy with intravenous golimumab.
Patients and methods
Patients
Adults with active RA despite MTX (stable regimen of 15–25 mg/week for ≥4 weeks) for ≥3 months were enrolled. Active RA was defined by ≥6/66 swollen joints and ≥6/68 tender joints at screening and baseline, rheumatoid factor positive and/or anticyclic citrullinated protein-positive at screening, and a screening C-reactive protein (CRP) concentration ≥1.0 mg/dl (upper limit of normal (ULN): 1.0 mg/dl). Patients were naïve to prior anti-TNF treatment. Stable (≥2 weeks) approved regimens of non-steroidal anti-inflammatory drugs and/or oral corticosteroids (≤10 mg/day) were allowed. Additional details of patient eligibility criteria are provided online.
Study design
The study (NCT00973479, EudraCT 2008-006064-11) was conducted according to the Declaration of Helsinki and the International Committee on Harmonisation good clinical practices. The protocol was reviewed and approved by each site's institutional review board or ethics committee. All patients provided written informed consent.
Patients enrolled in this multicentre, double-blind, placebo-controlled study were randomly (2:1) assigned, via interactive voice response system, to receive intravenous golimumab 2 mg/kg, or placebo infusions at weeks 0 and 4, and then q8 weeks through week 100, followed by 12 weeks of additional safety follow-up. Randomisation was stratified by screening CRP (<1.5 mg/dl, ≥1.5 mg/dl) and investigational site. Data collected at visits scheduled for weeks 0/2/4/8/12/14/16/20/24, are reported herein.
Patients randomised to placebo with <10% improvement in swollen/tender joint counts from baseline to week 16 early escaped (EE) from placebo to intravenous golimumab 2 mg/kg beginning with an induction regimen at weeks 16 and 20, followed by q8 week maintenance infusions. Patients assigned to golimumab also received placebo infusions at weeks 16 and 20 to maintain the study blind regardless of EE status; escalation beyond the 2 mg/kg dose was not permitted. All patients in both groups with <10% improvement underwent radiographic examination of the hands and feet at week 16.
Golimumab (Janssen Biotech Inc, Horsham, Pennsylvania, USA) was supplied in 4 ml vials of 12.5 mg/ml (50 mg/vial) and associated excipients; placebo infusions comprised 0.9% normal saline. Unless contraindicated, all study agent doses were infused over 30±10 min. All patients continued to receive a stable regimen of MTX (≥15 but ≤25 mg/week). Patients could receive standard prophylaxis for infusion reactions (eg, acetaminophen, antihistamines), but not corticosteroids, at the investigator's discretion.
The primary endpoint was the proportion of patients with an ACR20 response6 at week 14. Other measures of disease activity included determination of clinical response (a European League Against Rheumatism (EULAR) response of good or moderate response) and clinical remission (DAS28<2.6),7 ,8 both based on the disease activity composite score incorporating 28 joints and CRP (DAS28-CRP).9 ,10 Simplified Disease Activity Index (SDAI)11 and Clinical Disease Activity Index (CDAI)12 ,13 scores at weeks 14 and 24 were also calculated in a post hoc analysis. Decreases in these validated composite scores indicate reduced disease activity. Joint evaluations were performed by an independent blinded assessor assigned to each study centre. Improvement in functional status was assessed using the disability index of the health assessment questionnaire (HAQ).14 An improvement in the HAQ score ≥0.25 units was considered to be clinically significant, a level more stringent than previously deemed as meaningful improvement.15 Results of radiographic evaluations at weeks 0 and 24 and quality-of-life assessments will be reported separately. Blood samples collected at weeks 0 and 20 were evaluated for antibodies to golimumab using a bridging immunoassay.16 Safety endpoints included the incidence and type of adverse events (AEs), serious adverse events (SAEs), and incidence of infusion-related reactions, and changes from baseline in clinical laboratory parameters.
Statistical analyses
Patient data were summarised using descriptive statistics (counts/percentages for categorical data; median/IQR and mean±SD for continuous data). Statistical analyses were conducted by Janssen Research and Development LLC.
In the primary and major secondary efficacy analyses, proportions of patients achieving response levels were compared between treatment groups using a two-sided Cochran-Mantel-Haenszel test stratified by screening CRP (<1.5 mg/dl, ≥1.5 mg/dl). Additional prespecified analyses were also performed for week 0 (baseline) CRP cut-off levels of <1.0 mg/dl and ≥1.0 mg/dl. Patients with prohibited medication usage, who discontinued because of lack of efficacy before week 14 and who lacked all week 14 ACR20 component data for any reason were considered ACR20 non-responders at week 14 through week 24. In these intent-to-treat analyses, patients randomised to placebo who EE (n=68/197), and who received golimumab 2 mg/kg infusions at weeks 16 and 20, had week 16 data carried forward for response calculations at weeks 20 and 24. A last-observation-carried-forward procedure was employed to impute missing ACR component data (eg, swollen or tender joint count, or global assessments of disease) at week 14 if the patient had data for at least one other ACR component at week 14.
Sensitivity analyses of the primary endpoint included analyses in which patients who: (1) discontinued study agent due to an AE, (2) had insufficient data to determine ACR20 response and were considered ‘non-responders’, and (3) whose treatment regimen was inadvertently unblinded during the study were excluded. A fourth sensitivity analysis utilised a re-randomisation test. Additional treatment group comparisons of clinical response were accomplished via the χ2 or the Cochran-Mantel-Haenszel test (in cases involving stratification) for categorical variables and analyses of variance on the van der Waerden normal scores16 for continuous variables. All efficacy analyses were based on the intention-to-treat principle, and all statistical tests were performed at a two-sided significance level of 0.05.
Safety analyses were performed for all patients who received ≥1 infusion of study agent. Adverse event data were summarised as counts and percentages by treatment group and Medical Dictionary for Regulatory Activities (V.10.1) system-organ class and preferred term, and changes in laboratory parameters were summarised using descriptive statistics.
A sample size of 564 participants was estimated to provide ≥99% power to detect significant differences in treatment groups in the primary endpoint, assuming week 14 ACR20 response rates of ≥52% for golimumab, and 27% for placebo. The sample size also ensured an adequate number of patients exposed to intravenous golimumab 2 mg/kg for safety assessments, and provided sufficient power to address the major secondary endpoint (change from baseline in van der Heijde-modified Sharp score at week 24).
Results
Patient disposition and baseline characteristics
The first patient was screened on 14 Sept 2009; the last week 24 evaluation was performed on 18 May 2011. Among the 1219 patients screened, 592 were randomised. The study was conducted at 92 sites worldwide.
Baseline patient/disease characteristics were generally comparable between treatment groups (table 1). Most randomised patients were Caucasian (80.4%), and women constituted 81.6% of the population. Patients were 18–83 years of age (median: 52 years). At baseline, mean CRP concentration (2.6 mg/dl), DAS28-CRP score (5.9) and HAQ score (1.6) indicated substantial inflammation, severe disease activity and disability, respectively (table 1). The overall median disease duration was 4.7 years.
Summary of demographics at baseline; randomised patients
Among the 592 randomised patients, 570 (96.3%) completed the 24-week study period, and 22 (3.7%) discontinued study agent before week 24, most commonly because of AEs and the withdrawal of consent (figure 1). More than one-third (34.5%) of placebo+MTX-treated patients EE and initiated golimumab at week 16. Conversely, only 4.3% (n=17) of golimumab+MTX-randomised patients qualified for EE but did not change treatment (ie, 95.7% achieved ≥10% improvement in tender/swollen joint counts by week 16).

Patient disposition through week 24 of the GO-FURTHER trial. MTX, methotrexate.
Clinical response
The study's primary endpoint, that is, ACR20 response at week 14, was achieved, as 58.5% (231/395) of golimumab+MTX-randomised patients achieved an ACR20 response at week 14 versus 24.9% (49/197) of placebo+MTX-randomised patients (p<0.001, table 2). A significant difference in ACR20 response was observed as early as week 2 (33.2% (131/395) vs 11.7% (23/197); p<0.001 not corrected for multiplicity).
Summary of clinical efficacy among randomised patients. Data presented are number (%) of patients or mean±SD and median (IQR)
Results of all four sensitivity analyses comparing respective ACR20 response rates at week 14 yielded consistent results (p<0.001 for all comparisons) (table 2). Consistent treatment group differences in week 14 ACR20 response were also observed within the subgroups of patients with screening CRP <1.5 mg/dl, or ≥1.5 mg/dl, as well as within subgroups of patients with baseline CRP <1.0 mg/dl and ≥1.0 mg/dl (table 2). As shown in the supplemental online figure, significant treatment group differences were also observed across all subgroups of patients defined by baseline demographics and disease characteristics.
At week 14, a statistically significantly greater proportion (81.3%, p<0.001) of patients receiving golimumab+MTX demonstrated EULAR (moderate/good DAS28-CRP score) response versus placebo+MTX (40.1%, table 2). At week 2, DAS28-CRP response rates were 65.1% for golimumab+MTX and 19.3% for placebo+MTX (p<0.001 not corrected for multiplicity).
At week 14, patients in the golimumab+MTX group demonstrated significantly greater mean improvement in the HAQ score (0.50, p<0.001) versus placebo+MTX (0.19, table 2). At weeks 14 and 24, significantly higher proportions of patients had clinically significant improvement in the HAQ score (≥0.25 units) with golimumab+MTX (68.4% and 67.3%, respectively; p<0.001 at both timepoints) versus placebo+MTX (43.1% and 45.2%, respectively; table 2).
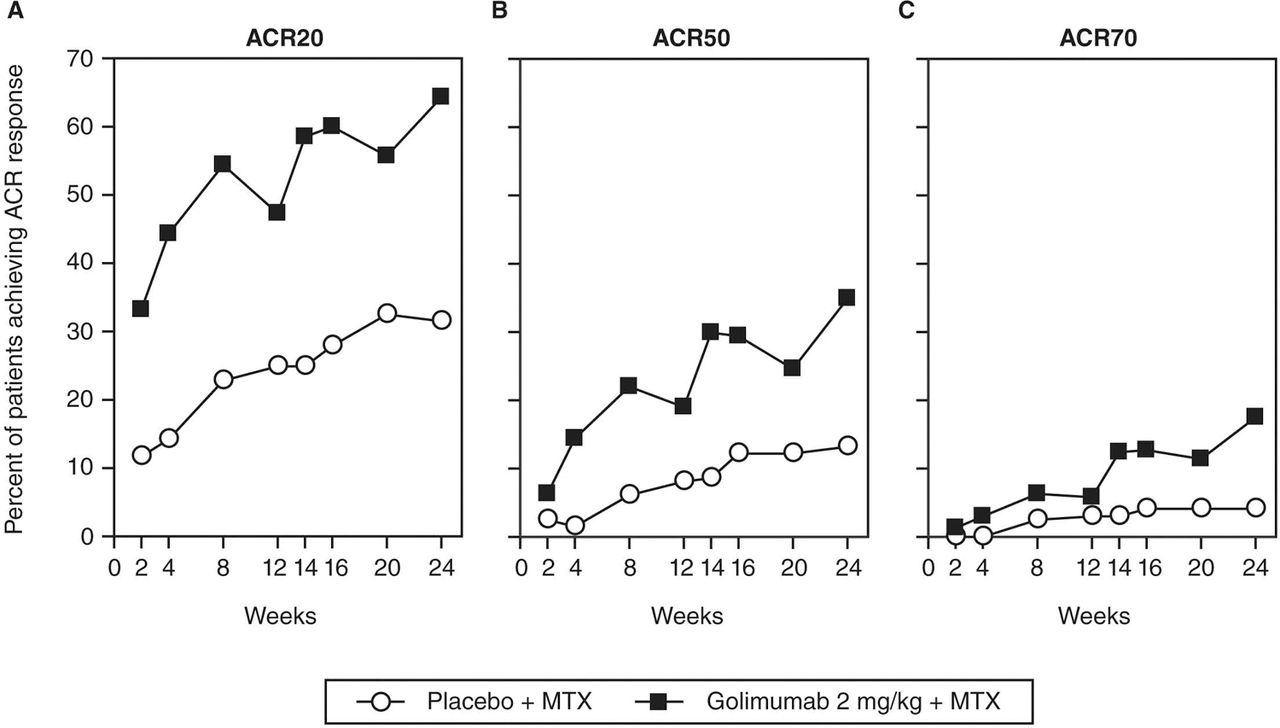
At week 24, the proportions of patients in the golimumab+MTX group achieving ACR50 (34.9%) and ACR70 (17.7%) responses were significantly greater than in the placebo+MTX group (13.2% and 4.1%, respectively; p<0.001 for both; table 2, figure 2). Among golimumab-treated patients, the proportion of ACR50 responders increased from 29.9% at week 14 to 34.9% at week 24 (figure 2). Similar patterns of improvement were also observed for ACR50 and ACR70 responses when based upon screening CRP ≥1.5 or <1.5 mg/dl and baseline CRP ≥1.0 and <1.0 mg/dl. Among golimumab-treated patients, the maximum improvement based upon ACR criteria tended to occur at week 24. There appeared to be small decreases in response rates at weeks 12 and 20 (ie, the presumed timing of trough serum golimumab concentrations) versus 4 weeks earlier (weeks 8 and 16, respectively; figure 2), although at all assessment timepoints from either week 2 forward (ACR20 (p<0.001) and ACR50 (p=0.05)) or beyond week 4 (ACR70, p=0.01), response rates were significantly higher with golimumab+MTX than with placebo+MTX.

Proportions of patients achieving ACR20 (A), ACR50 (B) and ACR70 (C) responses over time through week 24. ACR20, ACR50 and ACR70-American College of Rheumatology 20%, 50% and 70% response criteria, MTX, methotrexate.
Assessments of disease activity using the CDAI and SDAI composite indices also indicated significant improvement with golimumab+MTX (median improvements of 18.5 and 20.8, respectively) versus placebo+MTX (6.7 and 7.6, respectively; p<0.001 for both comparisons) at week 14. These improvements were sustained through week 24 (table 2). Disease activity categorised as remission/low/moderate/high showed similar trends (figure 3).

{kind=link}
{kind=link}
{kind=link}
Disease activity as assessed by the clinical disease activity index (CDAI) (A) and simplified disease activity index (SDAI) (B) composite scores at baseline, weeks 14 and 24. CDAI scores were categorised as remission (≤2.8), low disease activity (>2.8 and <10), moderate diesase activity (≥10 and <22) or severe disease activity (≥22), and SDAI scores were categorised as remission (≤3.3), low disease activity (>3.3 and <11), moderate diesase activity (≥11 and <26) or severe disease activity (≥26).13 p<0.001 for difference in distribution of weeks 14 and 24 scores among remission, low, moderate and high disease activity categories. GLM, golimumab; MTX, methotrexate.
Antibodies to golimumab
Antibodies to golimumab were detected in 13 (3.0%) of 440 golimumab-treated patients through week 24. Serum golimumab concentrations were generally lower in patients who tested positive versus negative for antibodies to golimumab (data not shown).
Safety
Similar proportions of placebo+MTX- and golimumab+MTX-treated patients reported AEs through week 16 (43.7% and 47.3%, respectively) and week 24 (49.2% and 52.9%, respectively). The most common AEs were ‘infections and infestations,’ including upper respiratory tract infection, urinary tract infection, and nasopharyngitis (table 3). Upper respiratory tract infection was the only AE reported in >5% of patients, with the exception of exacerbation of RA in the placebo+MTX group (5.6% of patients).
Summary of adverse events among treated patients through week 16 (placebo-controlled period) and week 24
During the 16-week placebo-controlled study period, AE reporting rates that were ≥1.0% higher in the golimumab+MTX than placebo+MTX group were observed for infections and infestations (24.3% vs 20.8%); nervous system disorders (6.8% vs 4.1%, respectively; mostly headaches); gastrointestinal disorders (6.6% vs 5.6%; predominantly diarrhoea and nausea); skin and subcutaneous tissue disorders (6.6% vs 3.6%; mostly pruritis and rash); respiratory, thoracic and mediastinal disorders (4.8% vs 2.5%, largely cough and dyspnoea); vascular disorders (3.8% vs 2.5%; predominantly hypertension); and metabolism and nutrition disorders (2.3% vs 0.0%). As would be expected in patients with RA receiving a less effective treatment regimen, a higher incidence of musculoskeletal disorders (largely RA) was observed in the placebo+MTX (14.7%) than golimumab+MTX (6.8%) group (table 3).
Through week 24, SAEs were observed in 2.0% and 4.1% of placebo+MTX- and golimumab+MTX-treated patients, respectively. Infections were the most common SAEs, reported by 0.9% (4/463) of golimumab+MTX- versus none of the placebo+MTX-treated patients. All SAEs were singular in nature, and no serious opportunistic or tuberculosis infections were reported. However, a non-serious oesophageal candidiasis infection was reported for one golimumab+MTX-treated patient.
Through week 24, one patient who received placebo+-MTX died of a presumed stroke due to hypertensive crisis. One treatment-emergent malignancy was reported through week 24: breast cancer in a 54-year-old woman (golimumab+MTX) at week 18. In addition, one case of non-treatment-emergent lung adenocarcinoma was reported in a 62-year-old woman (placebo+MTX) after week 4. No lymphoma or demyelination was reported through week 24.
Through weeks 24, 18 (1.1%) and 2 (0.2%), infusions were associated with an infusion reaction in patients numbering 16 (3.5%) and 1 (0.5%) in the combined golimumab+MTX and placebo+MTX groups, respectively. No severe or serious infusion reactions occurred. The median infusion time was 30 min, with 86.7% of infusions accomplished between 30 and 40 min. Approximately 5% (n=131) of placebo or golimumab infusions were completed within 20–30 min without any apparent increase in infusion reactions (data not shown).
In patients not receiving concomitant tuberculosis prophylaxis, and with baseline alanine aminotransferase (ALT) levels <ULN, a small difference was observed in the occurrence of ALT abnormalities between placebo+MTX (32/148, 21.6%) and combined golimumab+MTX (100/356, 28.1%) at week 24 (table 3). Two (0.6%) patients (golimumab+MTX) manifested ALT levels ≥5 times ULN. The remaining patients with elevations predominantly exhibited ALT levels >1 to <2 times ULN. In patients receiving tuberculosis prophylaxis, and with baseline ALT levels within normal limits, 20.6% (7/34) of placebo+MTX and 31.3% (20/64) of combined golimumab+MTX patients had ALT abnormalities through week 24.
Discussion
In the multicentre, double-blind, placebo-controlled GO-FURTHER study, patients with active RA despite background MTX therapy received intravenous golimumab 2 mg/kg, or placebo infusions, both in combination with stable MTX therapy, administered as a loading regimen at weeks 0 and 4 followed by q8-week dosing. At baseline, GO-FURTHER patients exhibited more inflammation (median CRP=1.9 mg/ml), greater disease severity per DAS28 scores, and more pronounced disability per HAQ scores than in previously conducted golimumab trials enrolling MTX-experienced patients with active RA (eg, median CRP=0.9 mg/dl in trials evaluating subcutaneous3 and intravenous5 golimumab). The degree of baseline active inflammation, disease severity and disability in the GO-FURTHER trial was actually more comparable with the ATTRACT study of infliximab (median CRP=2.6 mg/dl),17 which was conducted prior to the adoption of a revised approach to treating patients with RA, that is, at a time when clinical trials of RA cohorts had higher disease activity, especially in trials enrolling MTX-experienced patients.18
The study's primary endpoint (week 14 ACR20) was achieved, with a significant difference observed between the golimumab+MTX- and placebo+MTX-treated patients (58.5% vs 24.9%, p<0.001). The primary endpoint results were confirmed in four sensitivity analyses, as well as in subgroups of patients defined by screening/baseline CRP levels. Slightly more than one-third of golimumab+MTX-treated patients achieved an ACR50 response. Of note, in the previous intravenous golimumab trial, therapy was administered q12 weeks.5 The longer dosing interval in that trial yielded low systemic drug exposure in the later period of the 12-week dosing interval (weeks 8–12) and did not result in robust ACR50 response at early timepoints.
Also at week 14, a substantial and significantly greater proportion of patients in the golimumab+MTX group demonstrated an EULAR moderate/good response based on DAS28-CRP (81.3% vs 40.1%, p<0.001), with 65% of golimumab-treated patients achieving an EULAR response by week 2. Significant clinical improvement with golimumab was also evidenced when disease activity was assessed using the CDAI and SDAI composite scores. Importantly, these improvements in clinical signs and symptoms of RA were accompanied by improved physical function, with nearly 70% of golimumab+MTX-treated patients experiencing a clinically meaningful (≥0.25 units) improvement in the HAQ score15 at week 14.
Response to golimumab was rapid, with statistically significant treatment group differences in ACR20, ACR50, HAQ and DAS28-CRP responses observed by week 2, and in ACR70 response by week 4. More than 95% of golimumab-treated patients achieved ≥10% improvement in tender and swollen joints at week 16, compared with only 65% of placebo-treated patients.
The safety profile of intravenous golimumab was similar to that observed with intravenous placebo through week 16, and no new safety concerns were observed through week 24. Consistent with the safety of other anti-TNF agents,19 and with results of a previous trial of intravenous golimumab,5 infections were the most commonly reported AEs through week 24. Serious infections (4 events in 3 patients) were more commonly observed in patients receiving golimumab+MTX than placebo+MTX; no patient developed a serious opportunistic or tuberculosis infection. Through week 24, one patient (placebo+MTX) died of a presumed stroke at approximately week 22, and one treatment-emergent malignancy was reported (breast cancer, golimumab+MTX). No patient developed lymphoma, and no demyelination or congestive heart failure events were reported through week 24. Rates of infusion reactions were low, with reactions being isolated to a small number (13/395) of total golimumab-treated patients. Note that patients could receive standard infusion reaction prophylaxis (with the exception of corticosteroids) at the investigator's discretion, and there was no difference in rates of infusion reactions based upon whether patients did or did not receive such prophylaxis. Infusion reactions were largely mild or moderate, and none was life threatening. The median infusion time was 30 min, but approximately 89 of >2000 golimumab infusions were administered in 20–30 min with no evidence of increased rates of infusion reactions.
Overall, these results, through week 24 of the GO-FURTHER study, indicate that the addition of intravenous golimumab 2 mg/kg rapidly and significantly improved RA signs and symptoms and generally was well tolerated by patients with active RA despite ongoing MTX therapy. The GO-FURTHER trial continues through week 112, and further assessment of the benefit-risk profile of intravenous golimumab therapy in patients with RA is ongoing.
Acknowledgments
The authors thank Michelle Perate, MS, a paid consultant for Janssen Services, LLC, for writing assistance. The authors also thank the GO-FURTHER study investigators: Mohd Mokhtar Ainon, Kuantan, Malaysia; Kateryna Amosova, Kyiv, Ukraine; Evgenia Akatova, Moscow, Russia; Sang Cheol Bae, Seoul, Korea; Herbert SB Baraf, Wheaton, MD, USA; Vida Basijokiene, Siauliai, Lithuania; Alberto Berman, San Miguel De Tucuman, Argentina; Charles A Birbara, Worcester, MA, USA; Igor Bokarev, Moscow, Russia; David Hugh Bossingham, Cairns, Australia; Rodrigo Botero-Lopez, Medellin, Columbia; Jan Brzezicki, Elblag, Poland; Marek Brzosko, Szczecin, Poland; Loreta Bukauskiene, Klaipeda, Lithuania; J Michael Calmes, Lubbock, TX, USA; Daniel Wai Tho Ching, Timaru, New Zealand; Yun Yin Chong, Kota Kinabalu, Malaysia; Sook Khuan Chow, Selangor Darul Ehasan, Malaysia; Won Tae Chung, Pusan, Korea; Melvin A Churchill, Lincoln, NE, USA; Julian Alonso Coronel-Arroyo, Cali Valley Del Cauca, Columbia; Joana Dambrauskiene, Kaunas, Lithuania; Edmundo Hector de la Garza Ramos, Monterrey, Nuevo Leon, Mexico; Edit Drescher, Veszprem, Hungary; Anna Dudek, Warszawa, Poland; Alicia Eimon, Buenos Aires, Argentina; Hermann Leo Enkerlin-Pauwells, Durango, Mexico; Jorge Antonio Esquivel-Valerio, Monterrey, Mexico; Cecilio Flores-Garcia, Cozumel, Mexico; Andriy Gnilorybov, Donetsk, Ukraine; Peter James Gow, Hospital Road, Otahuhu, Auckland, New Zealand; Olena Grishyna, Kharkiv, Ukraine; Suk Chyn Gun, Seremban, Malaysia; Sergion Ramon Gutierrez-Urena, Guadalajara, Mexico; Stephen Hall, Melbourne, Australia; Joanna Hensel, Dzialdowo, Poland; Ihor Hospodarskyy, Ternopil, Ukraine; Heselynn Hussein, Precinct 7, Malaysia; Elena Ilivanova, Saint Petersburg, Russia; Juan Jose Jaller-Raad, Barranquilla, Columbia; Slawomir Jeka, Bydgoszcz, Poland; Young Mo Kang, Dae-Gu, Korea; Hyun Ah Kim, Anyang, Korea; Galyna Koshukova, Simferopol, Ukraine; Mikhail Kotelnikov, Moscow, Russia; Przemyslaw Kotyla, Katowice, Poland; Volodymyr Kovalenko, Kyiv, Ukraine; Virginija Kriauciuniene, Klaipeda, Lithuania; Anatoly Kuzin, Chelyabinsk, Russia; Maria Alicia Lazaro, Buenos Aires, Argentina; Pagalavan Letchumanan, Johor Bahru, Malaysia; Olena Levchenko, Odessa, Ukraine; Piotr Leszczynski, Poznan, Poland; Virginija Lietuvinikiene, Alytus, Lithuania; Mitchell B. Lowenstein, Palm Harbor, FL, USA; Michael Edmund Luggen Cincinnati OH, USA; Grygorii Lysenko, Kyiv, Ukraine; Daniel Augusto Machado, Rosario, Argentina; Maria Maciejowska-Roge, Wloszczowa, Poland; Marzin Mazurek, Lublin, Poland; Vadim Mazurov, St Petersburg, Russia; Paul Alejandro Mendez-Patarroyo, Bogota, Columbia; Michael F. Miniter, Moline, IL, USA; Jorge Luis Alberto Morales-Torres, Leon, Mexico; Jose Fernando Molina-Restrepo, Antioquia, Columbia; Lilla Nafradi, Szombathely, Hungary; Katalin Nagy, Eger, Hungary; Peter Thomas Nash, Maroochydore, Australia; Evgeny Nasonov, Moscow, Russia; Jacek Pazdur, Warszawa, Poland; Bernardo Antonio Pons-Estel, Santa Fe, Argentina; Rodolfo Ariel Pardo-Hidalgo, San Juan, Argentina; Won Park, InCheon, Korea; Yong Beom Park, Seoul, Korea; Margarita Pileckyte, Kaunas, Lithuania; Artur Racewicz, Bialystok, Poland; Oscar Luis Rillo, Buenos Aires, Argentina; Maureen Rischmueller, Woodville, SA, Australia; Zofia Ruzga, Wrocław, Poland; Pavel Shesternya, Krasnoyarsk, Russia; Yury Shvarts, Saratov, Russia; Seung Cheol Shim, Daejeon, Korea; Gagrath Pradeep (Joe) Singh, Takapuna, Auckland, New Zealand; Olga Smolenskaya, Ekaterinburg, Russia; Malgorzata Sochocka-Bykowska, Sopot, Poland; Mykola Stanislavchuk, Vinnitsa, Ukraine; Ingrid Strusberg, Cordoba, Argentina; Wahinuddin Sulaiman, Ipoh, Malaysia; Ferenc Szanyo, Gyor, Hungary; Istvan Szombati, Budapest, Hungary; Gabriella Szucs, Debrecen, Hungary; Malgorzata Szymanska, Warszawa, Poland; Bee Eng Tan, Georgetown, Malaysia; Guiiermo Alberto Tate, Buenos Aires, Argentina; Semen Ter-Vartanian, Kyiv, Ukraine; Cheng Lay The, Kuching, Malaysia; Yong H. Tsai, Daytona Beach, FL, USA; Phillip Vecchio, Woolloongabba, Australia; Algirdas Venalis, Vilnius, Lithuania; Natalia Vezikova, Petrozavodsk, Russia; Vadym Vizir, Zaporizhzhya, Ukraine; Michael A Weitz, Miami, FL, USA; Roman Yatsyshyn, Ivano-Frankovsk, Ukraine
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
-
Funding Janssen Research & Development LLC, and Merck/Schering-Plough provided support for this study.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.