Article Text

Abstract
Objective To test for a urate gene-by-diuretic interaction on incident gout.
Methods The Atherosclerosis Risk in Communities Study is a prospective population-based cohort of 15 792 participants recruited from four US communities (1987–1989). Participants with hypertension and available single nucleotide polymorphism (SNP) genotype data were included. A genetic urate score (GUS) was created from common urate-associated SNPs for eight genes. Gout incidence was self-reported. Using logistic regression, the authors estimated the adjusted OR of incident gout by diuretic use, stratified by GUS median.
Results Of 3524 participants with hypertension, 33% used a diuretic and 3.1% developed gout. The highest 9-year cumulative incidence of gout was in those with GUS above the median and taking a thiazide or loop diuretic (6.3%). Compared with no thiazide or loop diuretic use, their use was associated with an OR of 0.40 (95% CI 0.14 to 1.15) among those with a GUS below the median and 2.13 (95% CI 1.23 to 3.67) for those with GUS above the median; interaction p=0.006. When investigating the genes separately, SLC22A11 and SLC2A9 showed a significant interaction, consistent with the former encoding an organic anion/dicarboxylate exchanger, which mediates diuretic transport in the kidney.
Conclusions Participants who were genetically predisposed to hyperuricaemia were susceptible to developing gout when taking thiazide or loop diuretics, an effect not evident among those without a genetic predisposition. These findings argue for a potential benefit of genotyping individuals with hypertension to assess gout risk, relative in part to diuretic use.
Statistics from Altmetric.com
Gout is the most common inflammatory arthritis in the USA and its prevalence is increasing.1 Gout risk is mediated by both genetic and environmental factors. Hyperuricaemia is the strongest risk factor for gout,2 and previous studies have identified genes that influence serum urate concentrations.3,–,5 Of the eight genomic loci that were associated with serum urate levels in the Cohorts for Heart and Aging Research in Genome Epidemiology (CHARGE) consortium,3 five encode renal urate transporters or regulators thereof (SLC2A9, ABCG2, PDZK1, SLC22A11 and SLC17A1); the biological mechanism relating the other three (GCKR, R3HDM2-INHBC and RREB1) to elevated serum urate levels is unknown.
Although hyperuricaemia and gout have clear genetic determinants, there are additional environmental risk factors, such as the use of diuretic agents.6 Diuretics, particularly thiazides, lead to decreased renal clearance of urate due to increased reabsorption and are associated with an increase in serum urate levels and hyperuricaemia.7,–,10 In the Atherosclerosis Risk in Communities Study (ARIC), adults with hypertension who took thiazide or loop diuretics had an increased risk of developing gout over 9 years; this increased risk may be due to the increase in serum urate levels due to diuretic initiation.11 Diuretics share some of the tubular transport mechanisms for urate.12 Thus, there is biological evidence that a shared pathway may lead to diuretic-induced gout. We tested whether diuretic use is differentially associated with gout risk among participants with and without a genetic predisposition for elevated serum urate levels, such that there is a synergistic urate gene-by-drug interaction.
Materials and methods
ARIC is a prospective population-based cohort study of 15 792 individuals recruited from four US communities (Washington County, Maryland; Forsyth County, North Carolina; Jackson, Mississippi; and suburbs of Minneapolis, Minnesota, USA). The Institutional Review Board of the participating institutions approved the study protocol and participants provided written informed consent. ARIC recruited participants aged 45–64 in 1987–1989. This cohort was established to study the natural history of atherosclerosis, and consisted of one baseline visit between 1987 and 1989 and three follow-up visits conducted 3 years apart. ARIC is part of the CHARGE consortium that originally identified genes with a putative role in urate metabolism.3
We excluded participants who: were not Caucasian (n=4314); lacked available genotypes or did not consent to participate in genetic research (n=1736); did not report gout status (n=1910); or had prevalent gout at baseline (n=275). The study sample was limited to participants with hypertension (defined as on antihypertensive medication use or a measured blood pressure ≥140/90 mm Hg) at any visit (n=4033) to control for confounding by indication as was previously described.11
Trained interviewers collected medication information that participants used in the 2 weeks prior to each visit. We considered thiazide diuretics as a single class of diuretics and combined thiazide and loop diuretics (referred to as ‘thiazide or loop diuretic use’), as these two classes represent the majority of diuretic use and too few participants used loop diuretics (128 and 11 gout cases among those exposed to a loop diuretic). Any diuretic use additionally included potassium-sparing diuretics and other diuretics, not otherwise specified. The most common diuretic used was hydrochlorothiazide. The comparator groups were those not taking a diuretic (untreated hypertension as well as those who were treated with other antihypertensive treatments).
Genetic urate score (GUS) was calculated as published previously.3 The risk score included information on the single nucleotide polymorphism (SNP) with the strongest association with serum urate for each of eight genomic loci (rs2078267 in SLC22A11, rs780093 in GCKR, rs1106766 in R3HDM2-INHBC region, rs675209 in RREB1, rs1967017 in PDZK1, rs13129697 in SLC2A9, rs2199936 in ABCG2 and rs1165196 in SLC17A1) (table 1).3 For each SNP, the effect of the minor allele on serum urate from the published meta-analysis is multiplied by the number of minor allele copies in individual carriers; results are summed into a GUS. This score assigns the value of zero to participants who carry all major alleles and is reported in μmol/l.3 GUS represents the difference in mean serum urate levels of participants with a given combination of genotypes compared with those who carry two copies of the major alleles at all eight SNPs. A negative score suggests that the genetic profile of the participant is protective against elevated serum urate levels and a positive score suggests that the participant is at a genetically greater risk of elevated serum urate levels.
Information on urate genes
At the fourth visit, participants were asked, ‘Has a doctor ever told you that you had gout?’ and reported the age at gout diagnosis. The outcome was incident gout based on self-reported onset after baseline. Previous research suggests that self-reports of a physician diagnosis of gout are reliable (κ=0.73) and sensitive (sensitivity=84%).13
Serum urate concentrations were measured with the uricase method at visits 1 and 2 in mg/dl (convert to µmol/l by multiplying by 59.485). The reliability coefficient of serum urate was 0.91, and the coefficient of variation was 7.2% in a sample of 40 individuals with repeated measures taken at least 1 week apart.14 We scaled the mean serum urate level at visit 2 to account for lab drift.11
Other covariates of interest that were assessed at baseline (1989) included age, sex, body mass index (kg/m2) and alcohol use (grams/day). Serum creatinine was estimated using a modified kinetic Jaffé reaction. Glomerular filtration rate was estimated by using the CKD-EPI equation15 and categorised as ≥90, 60–90 or <60 ml/min/1.73 m2. These variables were considered as confounders of the interaction between urate handling genes and diuretic use because they were associated with diuretic use in this cohort.11
Using a logistic regression model, OR and 95% CI of incident gout for the use of a diuretic (thiazide diuretic, either a thiazide or loop diuretic, or any diuretic) compared with not using these treatments were estimated. These models were stratified by GUS median. Models were adjusted for confounders of the interaction of diuretic use and GUS on gout. The presence of effect modification of the association between diuretic use and incident gout by GUS was tested using a Wald test for the interaction term in the joint effects models. The interaction term was constructed by multiplying a dichotomous measure of genetic urate risk (above or below the median) and dichotomous diuretic use (present or absent) and this was added to the logistic model. We calculated the individual gene-by-diuretic interactions separately for the eight individual components of the genetic risk score. We tested for the presence of additional effect modification of GUS by diuretics by baseline serum urate level to account for the fact that patients with a higher GUS will have higher serum urate levels and present the OR for those with a genetic risk taking a diuretic and hyperuricaemia (>416 µmol/l). All statistical tests were considered to be significant at α<0.05.
Through sensitivity analyses, we tested whether there was a urate gene-by-drug interaction with non-diuretic antihypertensive treatments. Using a Cox Proportional Hazards Model, we estimated the GUS stratified HR of incident gout by diuretic use. We further adjusted the final logistic regression model for alcohol intake (grams/day or abstinence) and dietary factors (total calories, protein intake, vitamin C intake, fructose, and % calories from animal fat). All analyses were performed in SAS, V.9.1 (SAS Institute, Cary, North Carolina, USA).
Results
A total of 3524 ARIC participants with hypertension met the study criteria; 108 developed gout over 9 years (table 1). The 9-year cumulative incidence of gout was 3.1%; 1.8% in women and 4.5% in men. The study population was 47% male subjects. The mean (SD) age at cohort entry was 55 (5.6). There were 1179 (33%) participants taking any diuretic at any time during follow-up; 608 (17%) taking a thiazide; and 756 (21%) taking a thiazide or loop diuretic. The mean GUS was −1.15 μmol/l (SD=18.4; median score=−0.31). GUS was higher for participants who developed gout (−1.3 vs 4.9 μmol/l; p<0.001) and participants who developed gout while taking a diuretic (−1.7 vs 5.4 μmol/l; p=0.003).
Participants with a GUS above the median were more likely to be female subjects (55% vs 51%, p=0.01) (table 2). Participants who were female subjects, older age, obese or had low estimated glomerular filtration rate were more likely to have taken a diuretic (data not shown).
GUS, diuretics and incident gout
Baseline gout risk factors by median GUS in ARIC participants with hypertension (n=3524)
The 9-year cumulative incidence of gout was statistically higher among those who had GUS above the median and taking a diuretic compared with those who were not taking any diuretic (p=0.003 and p=0.002, respectively) (figure 1A). This effect was not evident for those with a GUS below the median.
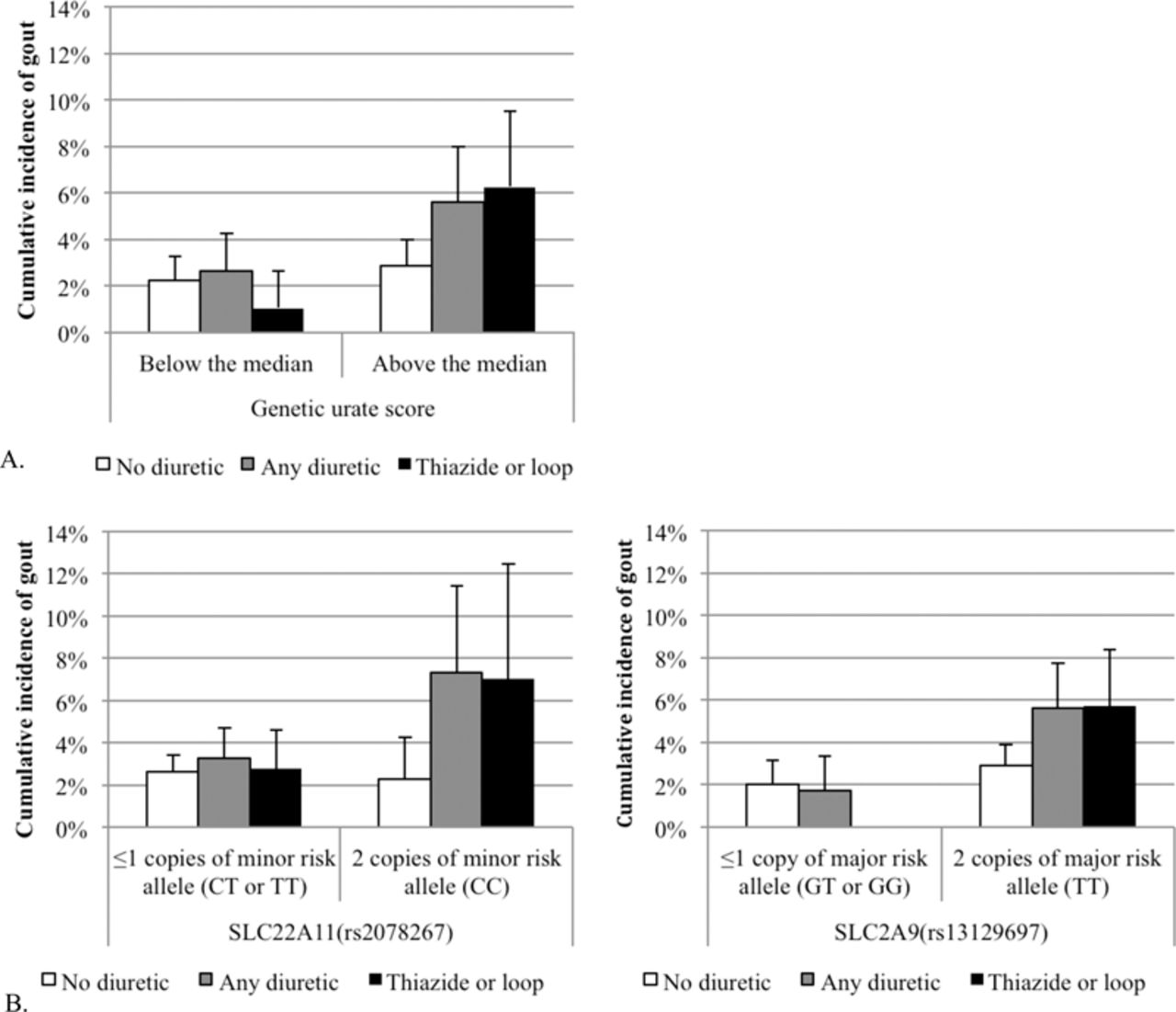
(A) Nine-year gout cumulative incidence by diuretic use (no diuretic use, any diuretic use, or thiazide or loop diuretic use) and median of genetic urate score (−0.31 μmol/l) in the Atherosclerosis Risk in Communities Study (ARIC) (n=3524); exact CIs. (B) Nine-year gout cumulative incidence by diuretic use and SLC22A11 (CC high risk genotype; frequency=21%) and SLC2A9 (TT high risk genotype; frequency=52%) in ARIC (n=3524); exact CIs. No participants carrying low risk alleles for SLC2A9 using either a thiazide or loop diuretics developed gout.
The adjusted OR of incident gout comparing those using a thiazide diuretic with those not taking a diuretic was 0.12 (95% CI 0.02 to 0.90) for individuals below GUS median, and 1.59 (95% CI 0.87 to 2.89) among those above the median (table 3). There was evidence of effect modification by thiazide diuretics (p=0.016). Furthermore, there was evidence of a urate gene-by-thiazide-by-urate three-way interaction (p=0.006) in a separate analysis. However, this analysis is limited because there was only one participant taking a thiazide diuretic who had GUS less than the median and developed gout.
Incident gout by diuretic use, stratified by median GUS in ARIC participants with hypertension (n=3524)
The adjusted OR of incident gout comparing those taking either thiazide or loop diuretics with no diuretic use was 0.40 (95% CI 0.14 to 1.15) for participants whose GUS was below the median and 2.13 (95% CI 1.23 to 3.67) for those with GUS above the median (table 3). This suggests there was evidence of effect modification (p=0.006). Furthermore, there was evidence of a three-way interaction among GUS, thiazide or loop diuretic, and baseline serum urate level (p=0.020) in a separate analysis. The 9-year adjusted OR of gout when taking a thiazide or loop diuretic among those with hyperuricaemia and a high genetic risk for elevated serum urate level was 1.97 (95% CI 1.06 to 3.67).
For individuals taking any diuretic, the adjusted OR of incident gout by diuretic use was 1.27 (95% CI 0.66 to 2.45) for those below the GUS median and 2.02 (95% CI 1.20 to 3.42) for those above the median, compared with those who were not taking a diuretic (table 3). There was no evidence for effect modification (table 3). Furthermore, there was little evidence of a urate gene-by-diuretic-by-urate three-way interaction (p=0.497) in separate analyses.
Figure 2 presents the predicted probability of gout by genetic risk and use of a thiazide or loop diuretic. Given individuals with the same serum urate level of 650 µmol/l, the risk of developing gout is 50% in those with a higher genetic risk and taking a thiazide or loop diuretic and 42% in those without an increased genetic risk and not taking a diuretic. The c-statistic for the full model was 0.82.

{kind=link}
{kind=link}
The predicted probability of developing gout over 9 years. D+, either thiazide or loop diuretic use; D−, no thiazide or loop diuretic use; G+, high genetic risk; G−, low genetic risk.
Individual SNPs, diuretics and gout
As not all of the urate-associated genes encode for renal urate transporters, the SNPs on which the GUS is based were also evaluated individually and two interactions were detected. Significant interactions for individual genetic effects with thiazide or loop diuretics were observed for rs2078267 in SLC22A11 (p=0.010) and for rs13129697 in SLC2A9 (p=0.010). The minor C allele of rs2078267 in SLC22A11 was associated with a higher serum urate. The cumulative incidences of gout by SLC22A11 and SLC2A9 carrier status are displayed in figure 1B.
Sensitivity analyses
There was no evidence of an interaction of other antihypertensive treatments with GUS (p>0.05). Using an adjusted Cox Proportional Hazards model, the results for thiazide (below median HR: 0.12, 95% CI 0.02 to 0.86; above the median HR: 1.44, 95% CI 0.81 to 2.57; p=0.018), either thiazide or loop diuretics (below median HR: 0.38, 95% CI 0.13 to 1.06; above the median HR: 1.76, 95% CI 1.04 to 2.88; p=0.009), and any diuretics (below median HR: 1.14, 95% CI 0.61 to 2.16; above the median HR: 1.73, 95% CI 1.04 to 2.88; p=0.340), were similar to the logistic model, suggesting that the results were robust to the statistical methods used and bias was not introduced by differential follow-up time. Adjusting for alcohol intake or dietary factors (total calories, per cent calories from animal fat, vitamin C intake and fructose intake) did not alter the significance.
Discussion
Our study demonstrates that White adults with a genetic risk for elevated serum urate may be at an increased gout risk when taking a thiazide or loop diuretic. Similarly, gout risk while taking a diuretic is not elevated and may be potentially decreased slightly in those with a low genetic risk. Ours is the first study to identify a urate gene-by-diuretic interaction on gout for a genetic risk score and specifically for individuals with urate-increasing variants at two of the genes summarised in the score, SLC22A11 and SLC2A9. Our results suggest that there is a synergistic effect between thiazide or loop diuretic use, high genetic risk for elevated serum urate levels, and elevated levels of measured serum urate on the risk of developing gout.
Decreased renal excretion of serum urate is thought to be the main cause of hyperuricaemia and gout.16 ,17 Renal excretion of urate is complex and depends on glomerular filtration as well as tubular secretion and reabsorption of urate. Anion exchange transport systems mediate urate flux and are present in the proximal tubular cells. Diuretics affect ion exchanger proteins in epithelial cells throughout the nephron. Chronic use of this preferred therapy for hypertension18 ,19 can lead to an increase in serum urate levels via increased urate reabsorption, which may be secondary to extracellular fluid volume contraction.20,–,22
In light of this, our findings for the SLC22A11 gene are of particular interest. SLC22A11 encodes OAT4, an organic anion transporter in the apical membrane of proximal tubule cells.23 OAT4 has been reported to provide an exit mechanism for loop diuretics into the tubular lumen (secretion) in exchange for urate and other filtered substrates into the proximal tubule cell (absorption).24 In support of the physiological importance of this finding, healthy individuals showed reduced fractional excretion of urate after the administration of the loop diuretic torasemide.24 It is conceivable that the exchange of diuretics for urate represents another mechanism by which the intake of loop diuretics may lead to increased serum urate concentrations and the increased incidence of gout observed in our population-based study. Certain genetic variants in SLC22A11 may lead to an increased transport capacity of OAT4, and the intake of diuretics may lead to an additional activation of the transporter, resulting in the highest incidence of gout in individuals with two copies of the urate-increasing allele and the intake of diuretics. In support of this theory, individuals with other genetic variants in SLC22A11 have been reported to show differences in the rate of bumetanide transport.25 However, the role of diuretics on GLUT9 transport has not been studied. Our study supports the interaction of urate genes and diuretic use; it was not designed to experimentally identify the exact biological mechanisms that explain why diuretic use is associated with gout among those with a genetic predisposition for elevated urate levels.
Some limitations of our study warrant mention. Gout was self-reported by participants; however, self-reported gout is both sensitive and reliable. It is unlikely that gout misclassification is differential to the presence of the interaction; thus, there is limited potential for bias due to misclassification.13 Additionally, we use the term ‘gene’ in the phrase ‘urate gene-by-drug interaction’ although we recognise that additional variants in each gene may be present and not modelled in this study. The biological mechanisms of GCKR, R3HDM2-INHBC and RREB1 with regard to elevated serum urate levels are currently unknown. We have still included these common alleles into the score in order to reflect the current knowledge of urate gene identification and to capitalise on the hypothesis-free nature of genome-wide association studies. The study collected information on diuretic use in the 2 weeks prior to the visit. However, antihypertensive treatments are often taken for years and patients refill and take this class of drugs on a regular basis.26 We had limited power due to a moderate sample size (n=3524 and 108 incident cases) to detect smaller interactions. Therefore, the main analysis focused on the GUS, which had more power to detect an interaction. This limited power may explain why the use of any diuretic was not significant, although there was a trend toward an interaction. The eight individual genes that were studied were defined a priori based on the genome-wide association studies and thus we did not correct for multiple testing. Furthermore, it is unlikely that the use of thiazide or loop diuretics is protective among those with a low genetic risk but deleterious among those with a high genetic risk; there were few participants who had a low genetic risk, exposed to a thiazide or loop diuretic and developed gout. As in all clinical research, it would be important to replicate these biologically plausible findings in multiple independent cohorts using parallel methods and include additional urate genes as they are identified.
To our knowledge, this is the first population-based study of diuretic use, urate-handling genes and incident gout in patients with hypertension. We found no evidence of effect modification by urate-handling genes on the association of other antihypertensive treatments and gout, suggesting that our results are specific to diuretics and not all antihypertension. Restricting our study population to those with hypertension allowed us to limit the confounding by diuretic indication. Although patients with hypertension are at a higher risk of developing gout,6 diuretic use is preferred therapy. Thus, patients with hypertension are the correct study source and target population.
Our results suggest that patients with a genetic risk of elevated urate, in particular those with at least two copies of the SLC22A11 minor allele (C; rs2078267), may want to be treated with antihypertensive treatments other than thiazide or loop diuretics. This association should be confirmed in additional studies before diuretic use becomes contraindicated in those who are genetically at risk for elevated urate levels. If diuretic use increases serum urate levels in all adults but only those with elevated genetic risk will go on to develop gout, then GUS may be useful for targeting diuretic treatment. However, the cost effectiveness of genetic testing should be studied prior to changing clinical practice. An algorithm that combines both genetic and environmental risk factors may be useful for gout risk assessment.
We identified a urate gene-by-diuretic interaction which increases the risk of developing gout. Use of a thiazide or loop diuretic in patients who are genetically predisposed to elevated serum urate may lead to the development of gout. We provide evidence for a potential mechanism, the OAT4-mediated exchange of diuretics and urate. Functional molecular studies that determine the biological mechanism of the gene-by-urate interaction would provide further evidence of a causal link between diuretic use and gout in those who are genetically predisposed to elevated urate levels.
Acknowledgments
The authors thank the staff and participants of the ARIC study for their important contributions.
References
Footnotes
-
Funding The Atherosclerosis Risk in Communities Study is carried out as a collaborative study supported by the National Heart, Lung, and Blood Institute contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C and HHSN268201100012C). Mara McAdams DeMarco was supported by a T32 training grant from the National Heart, Lung, and Blood Institute grant (5T32HL007024). Alan Baer and Janet Maynard were supported by the Donald B and Dorothy Stabler Foundation. Anna Köttgen was supported by the Emmy Noether Programme of the German Research Foundation. The funding sources had no role in the analysis or interpretation.
-
Competing interests None.
-
Ethics approval Approval provided by the Johns Hopkins Institutional Review Board.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement ARIC is available for public use through the NHLBI. This research was presented as an oral presentation at the American College for Rheumatology Annual Meeting in Chicago, Illinois, USA (7 November 2011).