Article Text

Abstract
In the last decade progress has been made in our understanding of bone biology. In particular, the relation between inflammation and bone has become much clearer, leading to bone-targeting therapies in inflammatory rheumatic diseases. The clinical sequelae of the influences of both inflammation and immobility (due to arthritis) on bone for different rheumatic diseases (such as rheumatoid arthritis, systemic lupus erythematosus and spondyloarthritides) have also now captured the attention of clinicians. In the last decade the well-known negative influences of glucocorticoids on bone have become more treatable as a result of new drugs that stimulate osteoblasts and restore the negative bone balance.
Statistics from Altmetric.com
Introduction
Bone is subject to a continuous remodelling process during life which allows the adaptation of our skeleton to individual demands. This process is based on the close interaction between the bone resorptive activity of osteoclasts and bone formation by osteoblasts, allowing a complete renewal of the adult skeleton roughly every 10 years. In children and adolescents bone formation exceeds bone resorption, allowing a gain in bone mass which reaches its maximum in early adulthood. In the adult skeleton bone resorption is in balance with bone formation, allowing the reshaping of the skeletal microarchitecture while maintaining a constant bone mass. With ageing, however, bone resorption usually exceeds bone formation, resulting in a net loss of bone mass which is associated with a greater fragility of the skeleton.
A number of factors can influence this bone remodelling process and disturb the natural balance between bone resorption and bone formation. Loss of oestrogens during the menopause, malnutrition associated with a poor calcium intake, excessive smoking or low physical activity such as in older patients in nursing homes are typical examples of conditions which lead to an imbalance in bone remodelling resulting in bone loss. Such factors are also of great relevance to the rheumatologist as diseases such as rheumatoid arthritis (RA), for instance, more frequently affect women in the menopause, are associated with smoking and often lead to impaired physical function due to pain and swelling of the joints. In addition, however, bone in patients with inflammatory rheumatic disease faces the assault of another major precipitator for bone loss—namely, inflammation itself.
Inflammation and bone
Two independent epidemiological studies indicated that even a small rise in the level of systemic inflammation can precipitate bone loss and emerge as an independent and strong risk factor for fractures.1 2 Bone loss thus shows striking parallels with cardiovascular disease; low-grade inflammation (as defined by a small but persistent elevation in the C reactive protein level) has not only been recognised as an independent risk factor for bone loss, but is also considered to be an established risk factor for cardiovascular disease. Moreover, bone loss and cardiovascular disease are both important long-term complications of chronic inflammatory rheumatic diseases such as RA, ankylosing spondylitis and systemic lupus erythematosus (SLE), and also of other types of chronic inflammatory diseases such as Crohn's disease. Given that the level of inflammation is substantially higher in chronic inflammatory disease than in low-grade inflammation, it is not surprising that the detrimental effects of inflammation on bone are manifesting far earlier and more pronouncedly in cases of chronic inflammatory disease.
There is no doubt that inflammation itself disturbs the balance between bone formation and bone resorption, triggering bone loss. How does this interaction work and which cells and molecules allow inflammation to play this influential role in disturbing homeostasis in our skeletal system? The clue to this question is immune activation which leads to the production of molecules with a negative effect on bone homeostasis. Chronic inflammation is usually characterised by the continuous activation of both the innate and adaptive immune system, both of which are also involved in inflammatory bone loss.
Enhanced osteoclastogenesis
Enhancement of osteoclast function is a key component of inflammatory bone loss. Osteoclasts are polykaryons derived from the monocyte lineage and thus belong to the innate immune system. Increased osteoclast activity during inflammation appears to result from a composite action of enhanced recruitment of osteoclast precursors from the bone marrow as well as induced differentiation of osteoclasts from monocytes. Cytokines such as tumour necrosis factor α (TNFα) trigger the evasion of osteoclast precursors into the bloodstream and to peripheral lymphatic organs and can thus enhance the systemic pool of osteoclast precursors.3 Increased differentiation of osteoclasts from their precursor cells appears to be another key component of inflammatory bone loss. Cytokines and prostaglandins support osteoclast differentiation by inducing the expression of key differentiation factors for osteoclasts. Expression of receptor activator of nuclear factor κB ligand (RANKL) and, to some extent, macrophage colony stimulating factor 1 are induced by cytokines involved in chronic inflammatory diseases such as the predominantly monocyte/macrophage-derived mediators TNF, interleukin 1 (IL-1) and IL-6, the stromal cell-derived cytokine IL-7 as well as the preferentially T cell-derived cytokine IL-17.4,–,6 In addition, ubiquitously expressed inflammatory mediators such as prostaglandin E2 can also induce the expression of RANKL and thus support osteoclast differentiation and bone resorption. The observation that lymphokines such as IL-17A can induce the expression of RANKL provides evidence that the adaptive immune system, in particular T cell activation, contributes to inflammatory bone loss through activation of osteoclasts.6 7 Moreover, T cells can act as a source of RANKL themselves and thus, in principle, can support osteoclastogenesis.8 9 The individual role of the different T cells on osteoclasts, however, might depend on their cytokine profile and the pattern of surface molecules they express. Thus, T helper 1 cells express RANKL but probably suppress osteoclastogenesis by their synthesis of interferon γ.10 Also, regulatory T cells express RANKL but potently suppress osteoclasts via their surface expression of cytotoxic T lymphocyte antigen 4, which inhibits osteoclast differentiation by direct cell to cell contact.11
Blunted osteoblastogenesis and osteocyte function
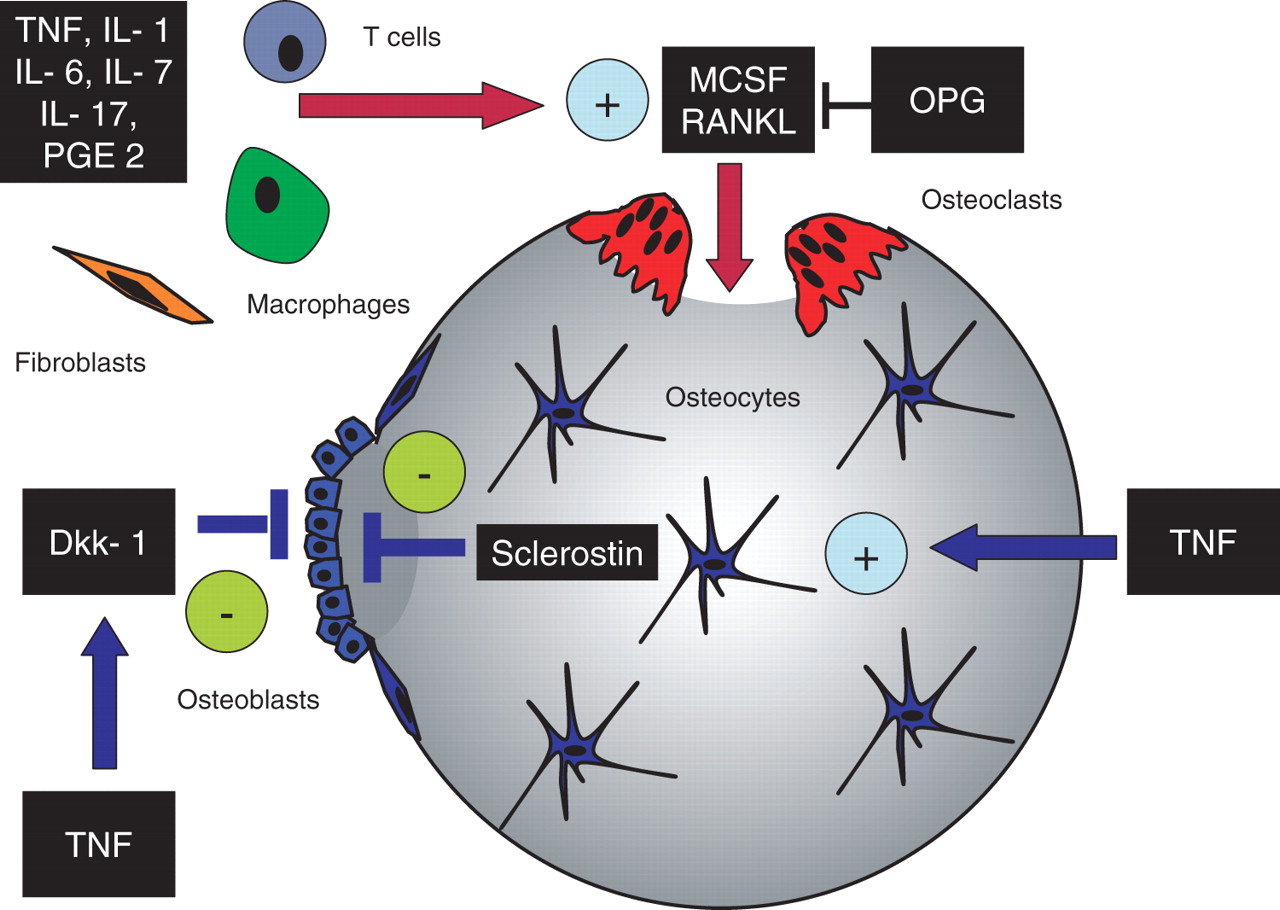
Given that enhanced osteoclastogenesis and increased bone resorption are key features of inflammatory bone loss, one would assume that increased bone formation could be compensatory. However, bone formation is blunted rather than enhanced in inflammatory disease. In physiological conditions, bone resorption is followed by adequate bone formation which restores the bone originally degraded by osteoclasts. This mechanism is also the principle of bone repair following micro- or macro-damage. Bone formation requires the differentiation and activation of osteoblasts, which are metabolically active cells originating from mesenchymal stem cells that synthesise bone matrix. The differentiation and activity of osteoblasts is regulated by a variety of molecules, of which parathyroid hormone (PTH), the bone morphogenic protein family including transforming growth factor β and the Wnt protein family are the most prominent factors. Inflammation can obviously interfere with the appropriate regulation of these signals and prevent the anabolic bone response usually elicited by osteoblasts. The factors of key relevance which allow the downregulation of bone anabolic pathways during chronic inflammatory disease have not yet been fully elucidated. TNFα, for instance, can induce the proteins Dickkopf 1 (DKK-1) and sclerostin, a product of the osteocyte, which are both potent inhibitors of the Wnt pathway and inhibit proper differentiation of osteoblasts and thus blunt bone formation.12 13 The levels of DKK-1 and sclerostin have been shown to control bone mass by influencing the activation state of the Wnt pathway. If induced by inflammatory signals, DKK-1 and sclerostin block the action of Wnt and prevent proper intracellular signalling leading to bone formation. This process not only blocks osteoblastogenesis but also indirectly affects bone resorption since the Wnt pathway controls the expression of osteoprotegerin which blocks the RANKL/RANK interaction and, as a consequence, also bone resorption. The shutdown of anabolic signals by inflammation can therefore be considered of key importance for the enhancement of bone resorption observed during inflammation, and could also explain the net gain of function of pro-osteoclastogenic molecules such as RANKL. Bone loss in chronic inflammatory disease, as summarised in figure 1, thus appears to result from low bone formation combined with enhanced bone resorption, which constitutes a detrimental imbalance of bone remodelling and precipitates the rapid loss of bone mass in the course of inflammatory disease.

Influence of inflammation on bone remodelling. Several inflammatory mediators such as tumour necrosis factor (TNF), interleukin (IL)-1, IL-6, IL-7, IL-17 and prostaglandin E2 (PGE2) which are produced by macrophages, T cells or fibroblasts stimulate osteoclast formation and bone resorption by inducing the expression of receptor activator of nuclear factor κB ligand (RANKL) and macrophage colony stimulating factor (MCSF). Both mediators are important for the differentiation and activation of osteoclasts. In addition, TNF induces Dickkopf 1 (Dkk-1), a Wnt antagonist, which blocks bone formation through inhibiting the differentiation of osteoblasts from mesenchymal precursors. Furthermore, TNF also induces the expression of sclerostin in osteocytes, which is a potent downregulator of bone formation. OPG, osteoprotegerin.
Effects of inflammatory arthritis on bone
The deleterious effect of inflammation on the quality of bone has been well documented in RA by local periarticular osteoporosis as well as by generalised osteoporosis.14 Generalised osteoporosis of the axial and appendicular skeleton in RA leads to an increased risk of fractures (adjusted RR for clinical osteoporotic fractures 1.5 (95% CI 1.4 to 1.6), most marked at the hip (RR 1.3) and spine (RR 2.4)) in patients without recent glucocorticoid (GC) use,15 but this increased risk is more than doubled in patients using GCs, even in low dosages (see below). In a detailed study in patients with early RA (as part of the BeSt study), changes in hand and in generalised bone mineral density (BMD) were evaluated during the first 2 years of treatment. A significant decrease in BMD was found in hands (most pronounced) and in the hip and spine. Interestingly, the more effective the treatment of RA, the less bone loss was observed.16 Studies based on bone biomarkers also confirm that more severe disease activity is associated with a higher rate of bone resorption.17 In different clinical studies it has now been suggested that blocking TNFα can indeed reduce bone resorption, which is accompanied by arresting the usually occurring bone loss in RA.18
Although patients with active inflammatory rheumatic disease have increased bone turnover leading to a heightened risk of fracture, there is no conclusive evidence that the threshold of BMD measurement to start treatment is different from that in postmenopausal osteoporosis. Apart from tight control of the underlying inflammatory disease, treatment modalities for established osteoporosis are not different from regular treatment modalities in postmenopausal osteoporosis. When patients are using GCs, specific treatment is indicated (see below).
The consequences of persistent inflammation in patients with juvenile idiopathic arthritis are comparable to those described in patients with RA.
In patients with spondyloarthritis (SpA) different mechanisms are involved, partly comparable to RA (predominantly leading to bone resorption) but partly unique to SpA, leading to new bone formation. Bone-inducing growth factors at entheseal sites may stimulate the development of dactylitis, syndesmophytes, ossification of ligaments and bony ankylosis in these patients.19 Although many patients with SpA have radiographic evidence of localised new bone formation, many of them have evidence of marked osteopenia and osteoporosis of the spinal column itself.20 An osteoporotic vertebral fracture in a patient with ankylosing spondylitis is not uncommon. Apart from inflammation itself, factors such as spinal immobility may, of course, play a role, although bone loss has been observed well before the development of spinal immobility. As in RA, treatment with TNFα blocking agents in SpA has been shown to improve skeletal remodelling.21
Reduced bone mass and osteoporosis in patients with SLE has been reported to be similar to that observed in patients with RA when controlled for age and disease duration.22 The effects of systemic inflammation, decreased physical activity, nutritional factors and drug treatment will all play a part in the observed decrease in systemic bone mass. In a study in over 100 patients with SLE (93% women, mean age 41 years), 20% of the patients had osteoporotic vertebral fractures.23 In this group, apart from the low body mass index and postmenopausal state, vitamin D deficiency was also found to be a relevant risk factor, possibly as a result of therapeutic advice on sun protection in patients with SLE.
Glucocorticoids and bone
Rheumatic diseases constitute the majority of health disorders for which GCs are used chronically.24 GCs affect bone by a decrease in bone formation mediated by osteoblast and osteocyte apoptosis together with increased bone resorption. Accelerated bone resorption is driven in part by RANKL (figure 2) and indirectly through reduction in sex hormones including oestradiol and testosterone.25 26 Of all these mechanisms, the direct effect of GCs on bone formation may be the most significant, as evidenced by histomorphometric studies showing reductions in osteoblast numbers and diminished trabecular wall width.27 Population-based data from the UK General Practice Research Database suggest that there is no clear safe dose of GCs with respect to bone.28 A heightened risk of fracture is seen even in those receiving subphysiological doses of GCs, and is further supported by a decline in biochemical markers of bone remodelling following only short-term use of oral, intra-articular or even inhaled GC therapy.29 The risk of fracture increases in a dose- and time-dependent fashion. In a study of patients with SLE, cumulative doses of GCs increased fracture risk more than average or peak dose.30 31 Fractures appear to develop in GC-induced osteoporosis partially independently of the effects of GCs on BMD, and an increased risk of fracture may occur as early as in the first few months of treatment.28 32 Trabecular bone is preferentially affected first at sites such as the spine and trochanter where losses of BMD average 3–10% during the first 6–12 months of GC therapy. Despite this pathogenic reasoning and accumulating evidence from observational studies on the risk of GCs to bone, some data suggest that GCs may actually benefit bones in patients with RA. This benefit may be brought about by decreasing disease activity, promoting beneficial weight-bearing activity and a reduction in proinflammatory cytokines deleterious to bone.33

{kind=link}
{kind=link}
Influence of glucocorticoids on bone remodelling. Summary of the many pathways by which glucocorticoids may influence bone quality, bone mass and fracture risk. Based on Canalis et al26 with permission. CSF, colony stimulating factor; GH, growth hormone; IGF, insulin-like growth factor; RANKL, receptor activator of nuclear factor κB ligand.
Prevention of GC-induced osteoporosis
Prevention of GC-induced osteoporosis begins with non-pharmacological lifestyle interventions such as increased weight-bearing physical activity, smoking cessation, mitigating the risk of falls and moderating alcohol/caffeine intake. Sufficient calcium and vitamin D are essential first steps for preserving bone and may be sufficient with careful serial monitoring and risk factor reassessment for very low-risk patients. International guidelines advocate 1500 mg elemental calcium (diet plus supplementation) and ≥800 IU vitamin D to help reduce the risk of fracture and offset urinary calcium losses caused by GCs. Activated forms of vitamin D such as calcitriol and alphacalcidiol have been used for prevention but are inferior to bisphosphonates in head-to-head trials with BMD end points.34,–,36 In some studies activated vitamin D was not significantly better at sustaining BMD than ergocalciferol.37 Based on well-designed clinical trials of patients with diverse diseases requiring GCs, the aminobisphosphonates alendronate, risedronate and zoledronate modestly increased bone mass or arrested bone loss in patients newly started on GCs (prevention studies) or those who were long-term users of GCs (treatment studies). While the studies on alendronate and risedronate involved a comparison with calcium and vitamin D alone as the placebo arm, zoledronic acid was tested against risedronate.38,–,41 Zoledronic acid given intravenously once yearly resulted in a greater increase in BMD at the spine and hip sites than risedronate.41 All studies were statistically underpowered to detect a reduction in the risk of fracture. However, in an extension of the original alendronate study, in a post hoc analysis of collapsed risedronate data (across a separate prevention and treatment study) and in a meta-analysis the reduction in the risk of vertebral fracture was of a similar magnitude to that seen in postmenopausal osteoporosis.42 An open-label study of intermittent intravenous ibandronate versus alphacalcidiol also demonstrated a reduction in fracture risk among those with established GC use.34
Studies of anabolic agents for the treatment of GC-induced osteoporosis have included testosterone and PTH analogues. There is a strong pathophysiological rationale for using these agents in GC-induced osteoporosis based on the known pathophysiology of the effects of GCs on bone. In a small crossover trial, testosterone was efficacious in improving bone mass in GC-induced osteoporosis.43 Teriparatide, a humanised recombinant 1–34 analogue of PTH, increased BMD at the spine and hip to a significantly greater extent than alendronate.44 In a secondary analysis of the teriparatide study, a lower rate of new vertebral spine fractures was seen with teriparatide compared with alendronate over a 3-year period of follow-up.45 The safety profile of bisphosphonates and anabolic agents in GC-induced osteoporosis studies has generally been very similar to that seen in other forms of osteoporosis. However, there was a significant increase in upper gastrointestinal events in the alendronate study and a slight rise in serum calcium in a small proportion of patients in the teriparatide study.38 44
Guidelines and treatment
International guidelines have predominantly advocated the use of calcium and vitamin D followed rapidly by bisphosphonates for chronic GC users.46,–,49 The use of sex steroids and calcitonin is supported by less robust data. Newer recommendations from the American College of Rheumatology now also indicate a potential role for teriparatide in patients at high risk for subsequent fractures on the basis of particularly low bone mass or a history of a prior fracture. Despite the growing evidence supporting a number of efficacious therapies to prevent and treat GC-induced osteoporosis, internationally many (if not most) at-risk persons are neither tested nor treated.50 51 Among those who receive anti-osteoporosis therapy, therapeutic effectiveness is limited by treatment persistence of <50% 1 year after starting treatment.52 The last decade has seen a considerable increase in our knowledge of the epidemiology, mechanisms and therapeutic approaches to GC-induced osteoporosis. Given the anticipated continued reliance by rheumatologists on GCs for many more years to come, better translation of evidence-based guidelines into clinical practice is needed to achieve improved public health in this important area of medicine.53
References
Footnotes
-
Provenance and peer review Commissioned; externally peer reviewed.
-
Competing interests None.