

Abstract
Objective. The role of natural killer (NK) cells in the immunopathogenesis of rheumatoid arthritis (RA) is unclear. Therefore, numerical and functional alterations of CD56dim and CD56bright NK cells in the early stages of RA development were studied.
Methods. Whole blood samples from newly diagnosed, treatment-naive, seropositive (SP) and seronegative (SN) patients with RA (SP RA, n = 45 and SN RA, n = 12), patients with SP arthralgia (n = 30), and healthy controls (HC, n = 41) were assessed for numbers and frequencies of T cells, B cells, and NK cells. SP status was defined as positive for anticyclic citrullinated peptide antibodies (anti-CCP) and/or rheumatoid factor (RF). Peripheral blood mononuclear cells were used for further analysis of NK cell phenotype and function.
Results. Total NK cell numbers were decreased in SP RA and SP arthralgia but not in SN RA. Also, NK cells from SP RA showed a decreased potency for interferon-γ (IFN-γ) production. A selective decrease of CD56dim, but not CD56bright, NK cells in SP RA and SP arthralgia was observed. This prompted investigation of CD16 (FcγRIIIa) triggering in NK cell apoptosis and cytokine expression. In vitro, CD16 triggering induced apoptosis of CD56dim but not CD56bright NK cells from HC. This apoptosis was augmented by adding interleukin 2 (IL-2). Also, CD16 triggering in the presence of IL-2 stimulated IFN-γ and tumor necrosis factor-α expression by CD56dim NK cells.
Conclusion. The decline of CD56dim NK cells in SP arthralgia and SP RA and the in vitro apoptosis of CD56dim NK cells upon CD16 triggering suggest a functional role of immunoglobulin G-containing autoantibody (anti-CCP and/or RF)-immune complexes in this process. Moreover, CD16-triggered cytokine production by CD56dim NK cells may contribute to systemic inflammation as seen in SP arthralgia and SP RA.
Rheumatoid arthritis (RA) is a chronic autoimmune disease. RA is manifested by inflammation of the synovial membrane mediated by joint-infiltrating immune cells. Increased expression of numerous cytokines and cytokine receptors has been observed early in the disease pathogenesis1,2,3,4. This poses a challenge for understanding the primary events in immune dysregulation involved in RA development. Current data support a role of interleukin 17 (IL-17) in the early phases of RA3,5. Despite the originally postulated pathogenicity of interferon-γ (IFN-γ), several reports demonstrated its protective role in the development of collagen-induced arthritis (CIA), a mouse model of RA6,7,8. The exact protective mechanism of IFN-γ in RA is currently not fully known9. Natural killer (NK) cells are primary IFN-γ producers9, by which they connect to the adaptive immune response and favor Th1 cell polarization in the course of an inflammatory response10,11,12. NK cell depletion was found to accelerate CIA onset, which was associated with an impaired IFN-γ–dependent regulation of the Th17 response7. Also, NK cells contribute to immune tolerance by killing autoreactive T cells and B cells13,14.
NK cells are divided into 2 major subsets based on the expression of CD56 (neural cell adhesion molecule)15. CD56dim NK cells, which constitute ∼90% of peripheral blood NK cells, are characterized by a potent cytotoxic capacity associated with increased perforin, granzyme, and cytolytic granule expression16. This suggests a primary role for CD56dim NK cells in the killing of autoreactive cells. CD56dim cells are more effective in antibody-dependent cellular cytoxicity when compared to the CD56bright subset, as a result of higher surface expression of FcγRIIIa (CD16). CD56bright NK cells are the minor subset (∼10%) within the circulating NK cell pool. However, in secondary lymphoid organs (e.g., lymph nodes17,18) and at several inflammatory sites (e.g., synovial fluid19, psoriatic plaques20) CD56bright NK cells have been shown to outnumber CD56dim cells. CD56bright NK cells may also have an immunoregulatory role owing to an increased ability (compared to CD56dim subset) to produce pro- and antiinflammatory cytokines15,16,21,22,23,24.
We aimed to investigate the role of NK cells in early stages of RA development, given their potentially initiating capacity in skewing and regulating the immune response. We studied newly diagnosed, treatment-naive patients with RA and patients with seropositive (SP) arthralgia. SP arthralgia is characterized by the presence of RA-associated autoantibodies [anticyclic citrullinated peptide antibodies (anti-CCP) and/or rheumatoid factor (RF)] and by tender and painful joints (arthralgia) but no synovitis. Previous studies show that 35% of patients with SP arthralgia develop RA after about 1 year of followup25. Thus, in the present study we investigated numerical and functional alterations of CD56dim and CD56bright NK cell subsets in patients with SP arthralgia and newly diagnosed RA.
MATERIALS AND METHODS
Patients
Thirty patients with SP arthralgia were defined based on seropositivity for RF (serum levels ≥ 15 IU/ml) and/or anti-CCP (serum levels ≥ 10 IU/ml), arthralgia in at least 1 joint, and lack of arthritis. Also, 45 patients with early RA SP for anti-CCP and/or RF, 12 patients with early seronegative (SN) RA (anti-CCP- and RF-), and 41 healthy controls (HC) were included in the study (Table 1). All patients with RA, fulfilling 1987 or 2010 American College of Rheumatology classification criteria for RA, were included in the study at the time of diagnosis, before start of treatment with disease-modifying antirheumatic drugs. Patients with SP arthralgia and RA received nonsteroidal antiinflammatory drugs (NSAID) only. HC were included only if, at the time of blood withdrawal, they had no infections, no recent vaccination, and did not use immunosuppressive drugs. All participants gave their written informed consent and the study was approved by the local medical ethics committee (University Medical Center Groningen, the Netherlands).
Demographic and clinical characteristics of the subjects included in the study.
Analysis of circulating leukocyte populations
Whole blood was analyzed using the BD MultiTest TruCount method with reagents detecting CD45, CD3, CD4, CD8, CD19, CD16/CD56, according to the manufacturer’s instructions (BD Biosciences). Flow cytometry was performed on FACS Canto II and analysis was performed using FACS Canto Clinical Software (BD Biosciences).
Analysis of NK cell phenotype and function
Heparin blood was used to isolate peripheral blood mononuclear cells (PBMC) by Lymphoprep (Axis-Shield) density gradient centrifugation, and PBMC were processed for cryopreservation. PBMC from all subjects were thawed at the same time and stained with the following antibodies: CD3 eFluor605NC, CD57 eF450 (eBioscience), CD56 FITC, CD16 Alexa Fluor700, CD94 APC, NKG2D PE-Cy7 (BioLegend), NKG2A PerCP (R&D Systems), and KIR2DL4 (Exbio Praha).
To assess NK cell IFN-γ expression, thawed PBMC were resuspended in RPMI-1640 containing 10% fetal bovine serum (FBS) and 0.6% gentamicin (Life Technologies) at a concentration of 106 cells/100 µl. Cells were incubated with phorbol myristate acetate (PMA) at a final concentration of 50 ng/ml, calcium ionophore at a final concentration of 1.6 µg/ml (both from Sigma-Aldrich), and BD GolgiPlu (BD Biosciences) diluted 1:1000. After 4 h at 37°C, PBMC were stained with the antibodies against CD3 eFluor605NC and CD56 PE (eBioscience). Cells were fixed and permeabilized with the Foxp3/Transcription Factor Staining Buffer Set (eBioscience) and stained with anti–IFN-γ Brilliant Violet 421 antibody (BioLegend).
To assess NK cell degranulation potency, analysis of CD107a was performed. Briefly, thawed PBMC were resuspended in RPMI-1640 with 10% FBS and 0.6% gentamycin at a concentration of 106 cells/100 µl. Cells were stimulated with PMA at the same concentrations as mentioned above in the presence of 0.5 µg of anti-CD107a Brilliant Violet 421 (BioLegend) antibody. After 1 h, BD GolgiPlug (diluted 1:1000) and BD GolgiStop (diluted 1:1000, both from BD Biosciences) were added and the stimulation was continued for another 5 h. After washing, PBMC were stained with antibodies: CD3 eFluor605NC and CD56 PE (eBioscience). PBMC were analyzed using an LSR II flow cytometer (BD Biosciences). Data analysis was performed with Kaluza analysis software (Beckman Coulter).
NK cell isolation and in vitro culture
For NK cell isolation and culture, blood from healthy volunteers (n = 6) was used and PBMC were isolated by Lymphoprep (Axis-Shield) density gradient centrifugation. PBMC were resuspended in PBS with 2 mM EDTA, 0.5% bovine serum albumin, and incubated with antibodies: CD3 eF450, CD56 PE, and CD19 PE-Cy7 (eBioscience). NK cell subsets CD3-CD19-CD56dim and CD3-CD19-CD56bright were isolated by fluorescence-activated cell sorting using MoFlo Astrios sorter (Beckman Coulter). Sorted CD56dim and CD56bright NK cells were resuspended in RPMI with 0.6% gentamicin and 5% FBS (Lonza) to a concentration of 5 × 105 cells/ml and incubated for 16 h at 37°C in 96-well flat-bottom polystyrene plates (Thermo Fisher Scientific). Culture conditions included 1000 U/ml human recombinant IL-2 (PeproTech), heat-aggregated rabbit immunoglobulin G (IgG; RAGG; Sigma-Aldrich) at a final concentration of 100 µg/ml, anti-CD16 antibody (clone 3G8; BioLegend) at a final concentration of 1 µg/ml, both IL-2 and RAGG, both IL-2 and anti-CD16, or medium alone. RAGG was prepared as described26.
Analysis of CD56dim and CD56bright NK cell apoptosis in vitro
After 16 h incubation, cell suspensions of sorted CD56dim and CD56bright NK cells were centrifuged, and supernatant was collected and stored at −20°C until analysis. Cell pellets were washed with PBS and resuspended with 3,3 -dihexyloxacarbocyanine iodide (DiOC6(3); Life Technologies) at a final concentration of 40 nM. After 15 min incubation at 37°C, cells were washed with PBS and analyzed immediately using an LSR II flow cytometer. Data analysis was performed with Kaluza analysis software.
Detection of cytokines in supernatants from cultured CD56dim and CD56bright NK cells
Levels of IFN-γ, tumor necrosis factor (TNF-α), IL-12, IL-4, IL-5, and IL-6 in the culture supernatants were quantified using Human Th1/Th2 Essential 6-plex Luminex assay (eBioscience) according to the manufacturer’s instructions. Data analysis was performed using StarStation software, version 2.3 (Applied Cytometry).
Statistical analysis
Statistical analysis was performed with GraphPad Prism version 5.0 (GraphPad Software) and IBM SPSS Statistics 20 (SPSS). Independent and dependent normally distributed variables were analyzed using ANOVA or repeated measures ANOVA, respectively, and Dunnett’s multiple comparison test (comparison against control). For non-normal, dependent samples a nonparametric Friedman test was performed, followed by Dunn’s posthoc test (comparison against control). P < 0.05 was considered statistically significant.
RESULTS
Seropositive patients, but not SN RA, are characterized by a decline in circulating NK cells
We aimed to identify peripheral immune alterations putatively involved in the early stages of RA pathogenesis and therefore compared the composition of the circulating lymphocyte pool (CD4+ T cells, CD8+ T cells, B cells, and NK cells) between patients (SP arthralgia, early SP RA, and early SN RA) and HC (Figure 1; Supplementary Figures 1 and 2, available online at jrheum.org). The number of total NK cells was significantly decreased in SP arthralgia and SP RA compared to HC (median 0.21 in SP arthralgia and 0.19 in SP RA vs 0.30 × 106 NK cells/ml in HC; Figure 1B). A similar decrease was observed for the proportions of NK cells within the total CD45+ pool (median 10.15% in SP arthralgia and 10.90% in SP RA vs 13.64% in HC; Supplementary Figure 1A, available online at jrheum.org). In contrast, the absolute numbers and the proportions of NK cells were not altered in patients with early SN RA (median 0.33 × 106 NK cells/ml and 19.31%, respectively). No significant alterations in the numbers of other circulating lymphocyte subsets were observed between patients and HC (Supplementary Figure 2, available online at jrheum.org).

Absolute numbers of CD56dim natural killer (NK) cells are decreased in seropositive (SP) arthralgia and SP rheumatoid arthritis (RA). A. Representative dot plots from a HC, an SP arthralgia patient, an SP RA patient, and an SN RA patient, showing the gating strategy to analyze CD56dim and CD56bright NK cell subsets within the total CD3-CD56+ NK cell population. B. Absolute numbers of total CD56+CD16+ NK cells in the blood of HC (n = 33), SP arthralgia patients (n = 30), early SP RA patients (n = 44), and SN RA patients (n = 11). C. The absolute numbers of CD56dim and (D) CD56bright NK cells were assessed using PBMC from HC (n = 32), SP arthralgia patients (n = 28), early SP RA patients (n = 43), and early SN RA patients (n = 10). The horizontal line in the scatter dot plots indicates the median; error bars indicate interquartile ranges. Statistical significance: * p < 0.05, ** p < 0.01. SN: seronegative; SAP: seropositive arthralgia patients; HC: healthy control; PBMC: peripheral blood mononuclear cells.
NK cells can be divided into 2 phenotypically and functionally distinct subsets based on CD56 expression (Figure 1A). The absolute numbers of CD56dim NK cells were decreased in both SP arthralgia (median 0.19 × 106 cells/ml) and SP RA (median 0.17 × 106 cells/ml) compared to HC (median 0.27 × 106 cells/ml; Figure 1C). The frequencies of CD56dim NK cells (within total CD3– cells), however, were not altered (Supplementary Figure 1B, available online at jrheum.org). In contrast, the absolute numbers of CD56bright NK cells were not different in SP arthralgia (median 0.014 × 106 cells/ml) or SP RA (median 0.018 × 106 cells/ml) when compared to HC (median 0.022 × 106 cells/ml). SN RA patients showed a specific increase of CD56bright NK cells in both their absolute numbers (median 0.040 × 106 cells/ml) and frequencies (median 4.44% of CD56bright cells within CD3–) when compared to HC (Figure 1D; Supplementary Figure 1C).
The mean age of patients with SN RA was higher than the mean age of the HC (Table 1). To exclude the possibility that the observed outcome is confounded by the age difference, a multiple linear regression analysis was performed. After adjusting for age, the previously observed differences in the absolute numbers and the frequencies of CD56bright NK cells between patients with SN RA and HC (p < 0.005 and p = 0.028, respectively) remained statistically significant (analysis not shown).
Next, we assessed whether the decline of NK cell numbers was associated with markers of general inflammation (C-reactive protein, erythrocyte sedimentation rate) or disease-specific characteristics (28-joint Disease Activity Score, anti-CCP, or RF level). We found a weak negative correlation between RF level and the absolute numbers of total NK cells (p = 0.034 and r = −0.23; data not shown).
NK cells from patients with SP RA showed decreased IFN-γ expression
Because we found the NK cell pool altered in SAP and patients with early SP RA, we next investigated their functionality by analyzing intracellular expression of CD107a and IFN-γ following PMA/Ca ionophore stimulation in vitro. Spontaneous expression of these markers did not differ among the groups (data not shown). NK cells from recently diagnosed patients with SP RA showed a decreased potency to produce IFN-γ compared to HC (median 51.4% vs 58.9% IFN-γ+ cells within CD3-CD56+ NK cells in SP RA and HC, respectively; Figure 2A). This was not observed for NK cells from SAP. When analyzing the contribution of the different NK cell subsets to the impaired capacity for IFN-γ production in SP RA, we noted that both the CD56dim and CD56bright subsets showed a reduced capacity for IFN-γ production in SP RA but not SP arthralgia (Figure 2C). Thus, the lesser capacity for IFN-γ production by NK cells from SP RA does not seem to be caused by the decline of CD56dim NK cells. No statistically significant differences in CD107a expression were observed between the studied groups (Figure 2B).

SP RA show decreased frequencies of IFN-γ+ NK cells. PBMC from HC (n = 8), SP arthralgia (n = 7), early SP RA (n = 9), and SN RA (n = 8) were stimulated for 4 h with PMA (50 ng/ml) and calcium ionophore (1.6 µg/ml). The frequency of (A) IFN-γ+ and (B) CD107a+ cells within total CD3-CD56+ NK cells (representative dot plots of 1 subject from each group are shown) and (C) the frequency of IFN-γ+ cells within subsets of CD56dim and CD56bright NK cells (representative dot plots from 1 HC are shown) was assessed. The horizontal line in the scatter dot plots indicates the median; error bars indicate interquartile ranges. Statistical significance: * p < 0.05. SN: seronegative; HC: healthy control; PBMC: peripheral blood mononuclear cells; SP: seropositive; SAP: seropositive arthralgia patients; RA: rheumatoid arthritis; IFN: interferon; NK: natural killer; PMA: phorbol myristate acetate.
NK cell function was also analyzed indirectly by assessing the surface expression of receptors with an activating (NKG2D, CD57), inhibitory (CD94/NKG2A), or activating/inhibitory role (KIR2DL4). In SP RA, significantly higher frequencies of NKG2D+ NK cells were found when compared to HC (median 74.1% vs 63.1% NKG2D+ cells within CD3-CD56+ NK cells in SP RA and HC, respectively). This was observed within the CD56dim NK cell subset only. No other differences in the frequencies of NKG2D+, CD57+, CD94/NKG2A+, or KIR2DL4+ NK cells between the studied groups were observed (Supplementary Figure 3, available online at jrheum.org). These data suggest an altered functionality of the peripheral NK cell pool in SP RA, but not in SP arthralgia.
CD56dim and CD56bright NK cell subsets: different susceptibility to FcγRIIIa-induced apoptosis
We next assessed whether the reduced NK cell numbers in SP arthralgia and SP RA might be explained by immune complex–mediated induction of NK cell apoptosis by FcγRIIIa (CD16) triggering. In line with prior studies, CD56dim NK cells demonstrated higher proportions of CD16+ cells than did CD56bright NK cells (Supplementary Figure 4). CD56dim and CD56bright NK cells were sorted from the blood of healthy volunteers (Figure 3A) and incubated with RAGG or agonistic anti-CD16 antibody in the presence or absence of recombinant IL-2. RAGG has been demonstrated to bind FcγR on NK cells and to mirror RF immune complexes27. Apoptosis was assessed by DiOC6(3) uptake analysis. Anti-CD16 alone and RAGG in the presence of IL-2 enhanced apoptosis of cultured CD56dim NK cells [median 14.14% and 21.89% DiOC6(3)low cells, respectively, Figure 3B] compared to the control [medium alone, 8.53% DiOC6(3)low cells]. IL-2 alone had no effect on the number of apoptotic CD56dim cells in vitro. Apoptosis of CD56dim NK cells by anti-CD16 antibody was further increased in the presence of IL-2 [35.06% DiOC6(3)low cells]. In contrast to CD56dim NK cells, anti-CD16, IL-2+anti-CD16, or IL-2+RAGG did not enhance apoptosis of CD56bright NK cells (Figure 3C).
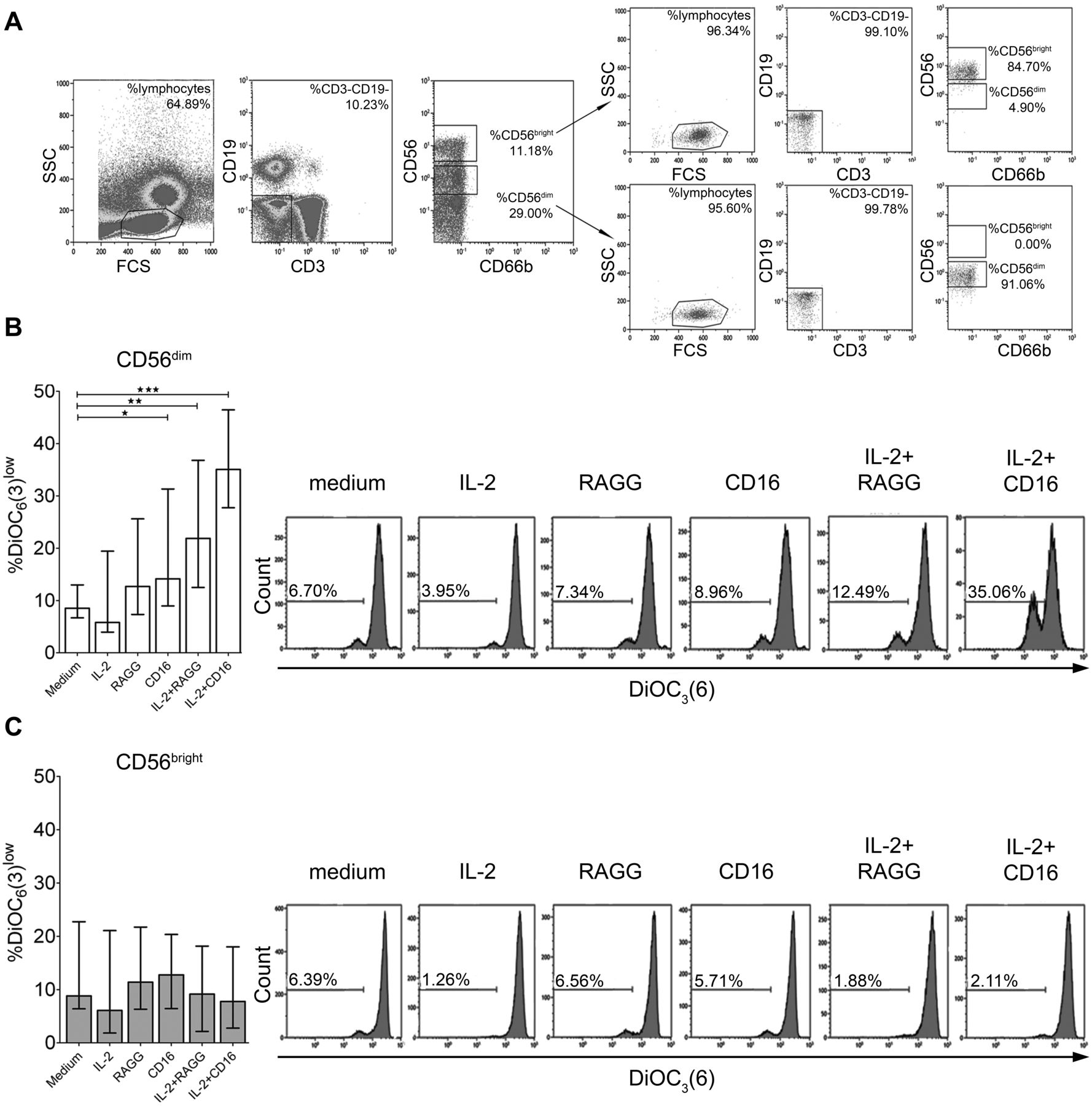
CD56dim but not CD56bright NK cells undergo apoptosis upon FcγRIIIa triggering. A. The gating strategy used in sorting for CD56dim and CD56bright NK cells and the reanalysis of the purity of sorted NK cell subsets are shown (median 85% and 96%, respectively). CD56dim and CD56bright NK cells from HC (n = 6) were cultured with IL-2, RAGG, anti-CD16, IL-2+RAGG, IL-2+anti-CD16, or medium alone. Frequencies of DiOC6(3)low cells within (B) CD56dim and (C) CD56bright events and representative histograms of 1 HC are shown. The horizontal line in the bar plots indicates the median; error bars indicate interquartile ranges. Statistical significance: * p < 0.05, ** p < 0.01, *** p < 0.001. HC: healthy control; NK: natural killer; IL: interleukin; RAGG: heat-aggregated rabbit immunoglobulin G; SSC: side scatter; FCS: forward scatter.
Our data show that CD56dim but not CD56bright NK cells undergo apoptosis upon FcγRIII-triggering in vitro, with the number of apoptotic cells increasing further upon the addition of IL-2.
CD56dim and CD56bright NK cell subsets: different propensity to produce IFN-γ and TNF-α following FcγRIIIa triggering
We next assessed the effect of CD16 triggering on cytokine production by NK cells from healthy donors. Sorted CD56dim and CD56bright NK cells (Figure 3A) were cultured with medium alone, IL-2, RAGG, anti-CD16, IL-2+RAGG, or IL-2+anti-CD16, and the supernatant was assessed for the production of IFN-γ, TNF-α, and other proinflammatory cytokines such as IL-12, IL-4, IL-5, and IL-6. In the presence of IL-2+RAGG or IL-2+anti-CD16, CD56dim NK cells produced IFN-γ and TNF-α (Figure 4A and Figure 4B). Increases in the production of IL-12 and IL-4 by CD56dim NK cells were also observed upon IL-2+RAGG culture (Figure 4C and Figure 4D). No statistically significant increase in cytokine expression was observed for CD56bright NK cell cultures (Figure 4).

CD56dim but not CD56bright NK cells produce IFN-γ, TNF-α, IL-12, and IL-4 upon FcγRIIIa triggering. CD56dim and CD56bright NK cells were sorted and cultured as described in Figure 3. After 16 h, supernatants from various culture conditions of CD56dim and CD56bright NK cells (from 4 HC used for Figure 3) were collected. Levels of (A) IFN-γ, (B) TNF-α, (C) IL-12, (D) IL-4, (E) IL-5, and (F) IL-6 were assessed using 6-plex cytokine assay. The horizontal line in the scatter dot plots indicates the median. Statistical significance: * p < 0.05, ** p < 0.01. HC: healthy control; NK: natural killer; IL: interleukin; RAGG: heat-aggregated rabbit immunoglobulin G; IFN: interferon; TNF: tumor necrosis factor.
DISCUSSION
We show a decline of NK cells in recently diagnosed RA and in patients with SP arthralgia, representing subjects at risk of progressing toward RA25. By stratifying our RA cohort according to autoantibody status, we found the NK cell decrease associated with SP but not SN RA. The decline in NK cells may be explained by a selective decrease of CD56dim NK cells as a result of apoptosis induction through FcγR triggering by IgG-containing immune complexes. In line with published data27,28,29,30,31,32, we observed the occurrence of FcγR-dependent (anti-CD16–induced) apoptosis of NK cells, augmented by IL-2, in vitro. We demonstrated differential susceptibility of CD56dim and CD56bright NK cell subsets to FcγR-induced cell death.
The decline of NK cells in SP arthralgia and in recently diagnosed SP RA suggests that this alteration may contribute to disease development rather than represent the consequence of longterm inflammation. Most of the published data describe similarly decreased NK cell numbers in later stages of RA33,34,35. The use of NSAID was found to have no effect on peripheral NK cell numbers36. Thus, NSAID are unlikely causal to the NK cell decline in SP arthralgia and SP RA.
Previously, NK cell depletion was found to accelerate the onset and augment the severity of CIA. Following the decline of NK cells, the decrease in systemic IFN-γ levels led to an expansion of Th17 cells directly involved in CIA induction. Further, the NK cell decrease was associated with plasma cell development and increased systemic levels of IgG autoantibodies7. This, together with the decline of NK cells in SP RA but not in SN RA, suggests a protective role for NK cells in the development of seropositive RA.
Despite similarly reduced NK cell numbers in SP arthralgia and early untreated SP RA, the decline of IFN-γ expression was observed in the latter group only. Thus, as shown for CIA7, the progression of pre-RA to overt disease may be associated with a reduction of NK cells as well as their functional impairment in production of IFN-γ. This would confirm the beneficial role of IFN-γ in arthritis pathology as shown in CIA6,7,8 and RA37.
The decline of peripheral NK cell numbers in SP but not in SN patients as well as the previously reported induction of NK cell apoptosis by FcγR triggering27,28,31 suggested a role of RA-related IgG-containing autoantibodies in this process. The majority of anti-CCP are IgG38 and can be bound by IgM RF39. A study by Boros, et al showed that IgM from RA sera was reactive with FcγRIII40. Further, about half of patients with RA have RF in a form of small IgG complexes41, which are efficiently bound by FcγRIIIa42.
We confirmed induction of NK cell apoptosis by agonistic anti-CD16 antibody and RAGG in vitro, a process that was enhanced by addition of IL-227,28,31. We observed a higher sensitivity of sorted CD56dim NK cells to FcγR crosslinking-induced apoptosis, which is likely the result of the higher expression of CD16 compared to CD56bright cells15 (Supplementary Figure 4, available online at jrheum.org). Differential susceptibility of CD56dim and CD56bright NK cells to FcγR-dependent apoptosis corresponds to the decline of circulating CD56dim but not CD56bright NK cells in the SP patients in vivo. It is unlikely that the decline of CD56dim NK cells in the periphery is a result of a preferential recruitment of this population to the joints because SF-derived NK cells were mainly of the CD56bright phenotype (data not shown19).
We observed an increased number of CD56bright NK cells in SN RA. Prior data suggest that the expansion of CD56bright NK cells is more specific for autoimmune diseases such as systemic lupus erythematosus43 or multiple sclerosis44 than RA and was associated with increased levels of type I interferons.
As shown29,30, CD16 triggering of CD56dim NK cells induced production of IFN-γ and TNF-α, cytokines implicated in RA pathogenesis. This process was augmented by the addition of IL-2. Similarly, we observed increased expression of Th1-specific (IL-12) and Th2-specific (IL-4) cytokines by CD56dim NK cells cultured with RAGG and IL-2. This suggests that CD56dim NK cells may be primarily responsible for the enhanced expression of a spectrum of various cytokines, although this feature has previously been attributed to CD56bright NK cells15,16,21,22,23,24. Increased IFN-γ and TNF-α expression by CD56bright compared to CD56dim was previously seen following stimulation with combinations of monocyte-derived cytokines21,22, PMA/ionomycin16,21, or whole bacterial pathogen24. Involvement of CD16 in the modulation of CD56dim NK cell cytokine expression has also been demonstrated45,46,47. Thus, proinflammatory cytokine production cannot be exclusively attributed to CD56bright NK cells. Depending on the available stimulus, both NK cell subsets can produce cytokines.
We also report that CD56dim NK cells in early SP RA are enriched with NKG2D+ cells compared to HC. Activating receptor NKG2D has been implicated in the development of autoimmunity48, and NKG2D blocking had beneficial effects on clinical, histological, and immunological variables (including reduction of IL-17 expression) in CIA49. This indicates that, next to the loss of protective function, CD56dim NK cells in SP RA may also display disease-promoting functions.
Our study focused on the systemic NK cell alterations, as it has been suggested that RA does not begin at the level of the joint but is preceded by systemic inflammation50. Our data suggest a role of circulating CD56dim NK cells in the pathogenesis of SP RA. On the other hand, enrichment of CD56bright NK cells in synovial fluid and tissue of longstanding RA19 indicates a role for these cells in the local inflammatory process in late-stage RA. However, it remains to be established whether CD56bright NK cells in the affected joints display inflammation-promoting or immunoregulatory properties.
We propose that the interaction between NK cells and RA-specific IgG-containing immune complexes is an early event in disease development. This is in line with the notion that the emergence of anti-CCP and RF autoantibodies occurs years before RA onset3. FcγRIIIa triggering of CD56dim NK cells by autoantibody-immune complexes could result in cytokine expression and a higher sensitivity to apoptosis of the CD56dim NK cell subset. The latter process is accelerated by IL-2, which, similar to ACPA and RF, was found increased in the periphery at the pre-RA stage3. Activation of CD56dim NK cells may thus contribute to early systemic inflammation as seen in SP arthralgia and SP RA. Moreover, the decline of CD56dim NK cells may allow for uncontrolled expansion of autoimmune cells contributing to RA development.
Also, our results demonstrate differences in the systemic immune profiles between SP and SN RA, adding to the notion that SP RA and SN RA may represent different disease entities.
ONLINE SUPPLEMENT
Supplementary data for this article are available online at jrheum.org.
Footnotes
Supported by the Groningen University Institute for Drug Exploration.
- Accepted for publication February 5, 2016.








{kind=link}
{kind=link}
{kind=link}
{kind=link}