Article Text

Abstract
Objectives To evaluate efficacy and safety of abatacept+methotrexate (MTX) in biologic-naive, anticitrullinated protein antibody (ACPA)-positive Japanese patients with active rheumatoid arthritis (RA) and early erosion versus placebo+MTX.
Methods In this phase IV, multicentre, double-blind study (NCT01758198), patients were randomised (1:1) to receive intravenous abatacept (~10 mg/kg) or placebo, plus MTX (≥6 mg/week). Primary efficacy objectives were to compare American College of Rheumatology 20 (ACR20) response rates at week 16 and mean change from baseline in van der Heijde-modified total Sharp score (vdH-mTSS) at week 24 between abatacept+MTX and placebo+MTX groups.
Results Overall, 203 and 202 patients received abatacept+MTX and placebo+MTX, respectively. At week 16, ACR20 response rates were higher in the abatacept (75.4%) versus placebo group (27.7%; p<0.001). Mean change from baseline in vdH-mTSS at week 24 was 0.84 in the abatacept and 1.26 in the placebo group (p=0.017). Radiographic non-progression rates (change in vdH-mTSS≤smallest detectable change (1.9)) were 88.1% and 75.4% in abatacept and placebo groups, respectively. Adjusted mean change from baseline in Disease Activity Score 28 (C-reactive protein) (DAS28 (CRP)) at week 16 demonstrated a numerically greater reduction in the abatacept versus placebo group. Proportions of patients with DAS28 (CRP), Simplified Disease Activity Index and Clinical Disease Activity Index remission up to week 52 were higher in the abatacept versus placebo group. The abatacept safety profile was consistent with previous observations.
Conclusions Compared with MTX alone, abatacept+MTX improved clinical symptoms and inhibited structural damage progression in ACPA-positive, Japanese patients with RA, early erosion and inadequate response to MTX.
- DAS28
- disease activity
- rheumatoid arthritis
- DMARDs (biologic)
- Anti-CCP
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Previous studies have demonstrated that a high proportion of Japanese patients with rheumatoid arthritis (RA) are anticitrullinated protein antibody (ACPA) positive.
Abatacept, a co-stimulatory signal inhibitor, was approved in Japan in 2010 for the treatment of patients with RA who had an inadequate response to previous conventional synthetic disease-modifying antirheumatic drugs.
What does this study add?
This randomised, double-blind, placebo-controlled phase IV study demonstrated that abatacept with methotrexate (MTX) improved clinical symptoms and inhibited structural damage progression in ACPA-positive Japanese patients with early erosive RA and an inadequate response to MTX alone.
How might this impact on clinical practice?
This study suggests that abatacept with MTX could be considered a suitable treatment option for biologic-naive Japanese patients with RA.
Introduction
Rheumatoid arthritis (RA) is a chronic, inflammatory autoimmune disease characterised by synovitis leading to progressive joint damage and subsequent impaired physical function and which requires long-term management.1 Poor prognostic factors (PPFs) for RA include the presence of rheumatoid factor (RF) and/or anticitrullinated protein antibody (ACPA) or early erosions.1 The addition of biologic disease-modifying antirheumatic drugs (bDMARDs) or targeted synthetic DMARDs (tsDMARDs) to treatment with conventional synthetic DMARDs (csDMARDs) is recommended for patients with PPFs who responded inadequately to previous csDMARDs.2
Abatacept is a human fusion protein that selectively modulates the CD28–CD80/CD86 co-stimulatory signal required for full T-cell activation.3 In Japan, abatacept was approved in 2010 for the treatment of patients with RA who had an inadequate response to previous csDMARDs. To investigate safety and clinical efficacy, phase II and III studies of abatacept in Japanese patients with RA were conducted.4–6 However, none of these studies investigated efficacy on joint protection or focused on those patients with PPFs. Previous studies have demonstrated that a high proportion of Japanese patients with RA are ACPA positive (79.2%–86.6%)7–10; thus, efficacy and safety of bDMARDs or tsDMARDs with concomitant use of methotrexate (MTX) are of interest in this patient population.
In this study, the efficacy and safety of abatacept with concomitant use of MTX were investigated in a randomised, double-blind and placebo-controlled phase IV trial in biologic-naive, ACPA-positive patients with active RA and early erosion.
Methods
Study population
Biologic-naive and ACPA-positive patients aged ≥20 years with active RA (defined by American College of Rheumatology (ACR) 198711/European League Against Rheumatism (EULAR) 2010 criteria12) for ≥16 weeks prior to screening were eligible for inclusion. In addition, eligible patients were <5 years from diagnosis and had received MTX (≥6 mg/week) for ≥3 months. Eligible patients also had ≥6 swollen joints among 66 screened, ≥6 tender joints among 68 screened, investigator-diagnosed erosion and C-reactive protein (CRP) ≥2.0 mg/dL or erythrocyte sedimentation rate (ESR) ≥28 mm/hour. All patients provided written informed consent prior to participation in this study.
Patients were excluded if they met the diagnostic criteria for any other rheumatic disease such as systemic lupus erythematosus (those with Sjögren’s syndrome, osteoarthritis, gout and osteoporosis were eligible). Further exclusion criteria are detailed in the online supplementary material.
Supplemental material
Study design and treatment
This phase IV, randomised, double-blind, placebo-controlled, 52-week observation, postmarketing study (ClinicalTrials.gov identifier, NCT01758198) was conducted between 24 April 2013 and 26 December 2016 at 71 sites in Japan.
After screening, eligible patients were randomised (1:1; without stratification) to receive intravenous abatacept plus MTX (≥6 mg/week) or matched intravenous placebo plus MTX (≥6 mg/week). During the study period, patients could receive MTX continuously at the study initiation dose. Abatacept was administered by intravenous infusion at the dose approved in Japan in accordance with the patients’ body weight (<60 kg: 500 mg; ≥60 to ≤100 kg: 750 mg; >100 kg: 1000 mg) on day 1, at weeks 2 and 4 and every 4 weeks thereafter for up to 48 weeks. Both investigator and patient remained blinded throughout the study. Randomisation and allowed concomitant medications are detailed in the online supplementary material.
Patients who did not achieve a ≥20% improvement from baseline in swollen and tender joint counts after the evaluation at week 16 were offered open-label intravenous abatacept (~10 mg/kg) every 4 weeks as rescue therapy, regardless of the original randomisation arm allocation and without breaking the randomisation blind. However, the final decision regarding use of abatacept rescue therapy was determined by the investigator.
Disease activity and physical function assessments
Disease activity was evaluated using ACR response rates,13 Disease Activity Score 28 (CRP) (DAS28 (CRP)),14 15 Simplified Disease Activity Index (SDAI) and Clinical Disease Activity Index (CDAI).16 The proportion of patients achieving ACR20 response at week 16 was one of two co-primary efficacy endpoints. ACR50 and ACR70 response rates and the change from baseline in DAS28 (CRP) at week 16 were secondary efficacy endpoints. Tertiary efficacy endpoints included the proportion of patients with remission defined in this study by DAS28 (CRP) <2.6, SDAI≤3.3 or CDAI ≤2.816 at weeks 16, 24 and 52. Physical function was assessed using the Japanese version17 of the Health Assessment Questionnaire-Disability Index (HAQ-DI).18 HAQ-DI response rates, defined as a ≥0.3 reduction from baseline, were evaluated at week 24 and 52 as tertiary efficacy endpoints.
Radiographic assessment
Joint damage progression was evaluated by van der Heijde-modified total Sharp score (vdH-mTSS) method for scoring radiographs of hands and feet in RA including 16 areas in each hand for erosions and 15 for joint-space narrowing (JSN).19 The change from baseline in vdH-mTSS at week 24 was the second co-primary efficacy endpoint. The rate of radiographic non-progression at week 24, defined as the proportion of patients with a change from baseline vdH-mTSS ≤1.9 (smallest detectable change (SDC)),20 was a secondary endpoint. Tertiary efficacy endpoints are detailed in the online supplementary material.
Safety assessment
Safety was assessed throughout the study by recording the incidence of all adverse events (AEs) or serious AEs (SAEs), discontinuations due to AEs and AEs of special interest including those associated with immunomodulatory drug use, infections, prespecified autoimmune disorders, malignancies and infusional reactions (definitions detailed in the online supplementary material). Further details on AE classification are presented in the online supplementary material.
Statistical analyses
The study sample size of 400 patients was determined based on the following considerations. Assuming a placebo response rate of 20%, a difference of 45% in the ACR20 response rate can be detected with at least 99% power at a significance level of 5% (two-sided). Based on the hierarchical testing procedure used for the co-primary endpoints, the difference of 1.9 (assuming SD 5.5) in the change from baseline in mTSS using the vdH-S method can be detected with >90% power at a significance level of 5% (two-sided), using a Wilcoxon rank sum test. Patients who received rescue abatacept treatment or withdrew for any reason were considered non-responders from that time point onwards. Efficacy analyses were performed in the intent-to-treat population which included all randomised patients who received at least one dose of study medication. The differences in ACR20 response rates between treatment groups at week 16 were analysed by χ2 test with continuity correction. Change from baseline in vdH-mTSS at week 24 was compared using a rank-based non-parametric analysis of covariance model, with the rank score for change from baseline in vdH-mTSS as the dependent variable, treatment group as a main effect and the rank score for baseline in vdH-mTSS as a covariate. Further statistical analyses are detailed in the online supplementary material.
Results
Patient disposition and baseline characteristics
Patient disposition is shown in figure 1. Of 547 patients enrolled, 406 were randomised to treatment and 405 received at least one dose of the study drug; abatacept plus MTX group (n=203) and placebo plus MTX group (n=202). Baseline demographics, disease characteristics, treatment history and concurrent medication were comparable between the two treatment groups (table 1). Over the study period, a total of 29 patients discontinued; 12 (three abatacept plus MTX; nine placebo plus MTX) before week 16; five (two abatacept plus MTX; three placebo plus MTX) between weeks 16 and 24 and 12 (six abatacept plus MTX; six placebo plus MTX) between weeks 24 and 52. These discontinuations were mostly due to AEs (seven (3.4%) abatacept plus MTX; six (3.0%) placebo plus MTX) and lack of efficacy (two (1.0%) abatacept plus MTX; eight (4.0%) placebo plus MTX). AEs that led to discontinuation in the abatacept plus MTX group included interstitial lung disease (n=2), lymphoma, erythema nodosum, viral infection, colon cancer and abnormal liver function test. In the placebo plus MTX group, AEs that lead to discontinuation included suicide, Pneumocystis jirovecii pneumonia, drug hypersensitivity, anaphylactic shock, tendon rupture and drug-induced liver injury. Abatacept rescue therapy was received by 25 (12.3%) patients in the abatacept plus MTX group between week 16 and 24 and by 12 patients (5.9%) between week 25 and 52. In contrast, 116 (57.4%) patients in the placebo plus MTX group received rescue therapy between week 16 and 24 and 22 (10.9%) between week 25 and 52 (figure 1).
Patient demographics and baseline characteristics

Patient disposition over the 52-week study period. *Patients did not meet study inclusion criteria (patient may have more than one reason), including 62 with CRP <2.0 mg/dL or ESR <28 mm/hour ESR, 26 ACPA-negative and 20 with ALT >2×ULN. ACPA, anti-citrullinated protein antibody; ALT, alanine transaminase; CRP, C-reactive protein; ESR, erythrocyte sedimentation rate; MTX, methotrexate; ULN, upper limit of normal.
Disease activity and physical function
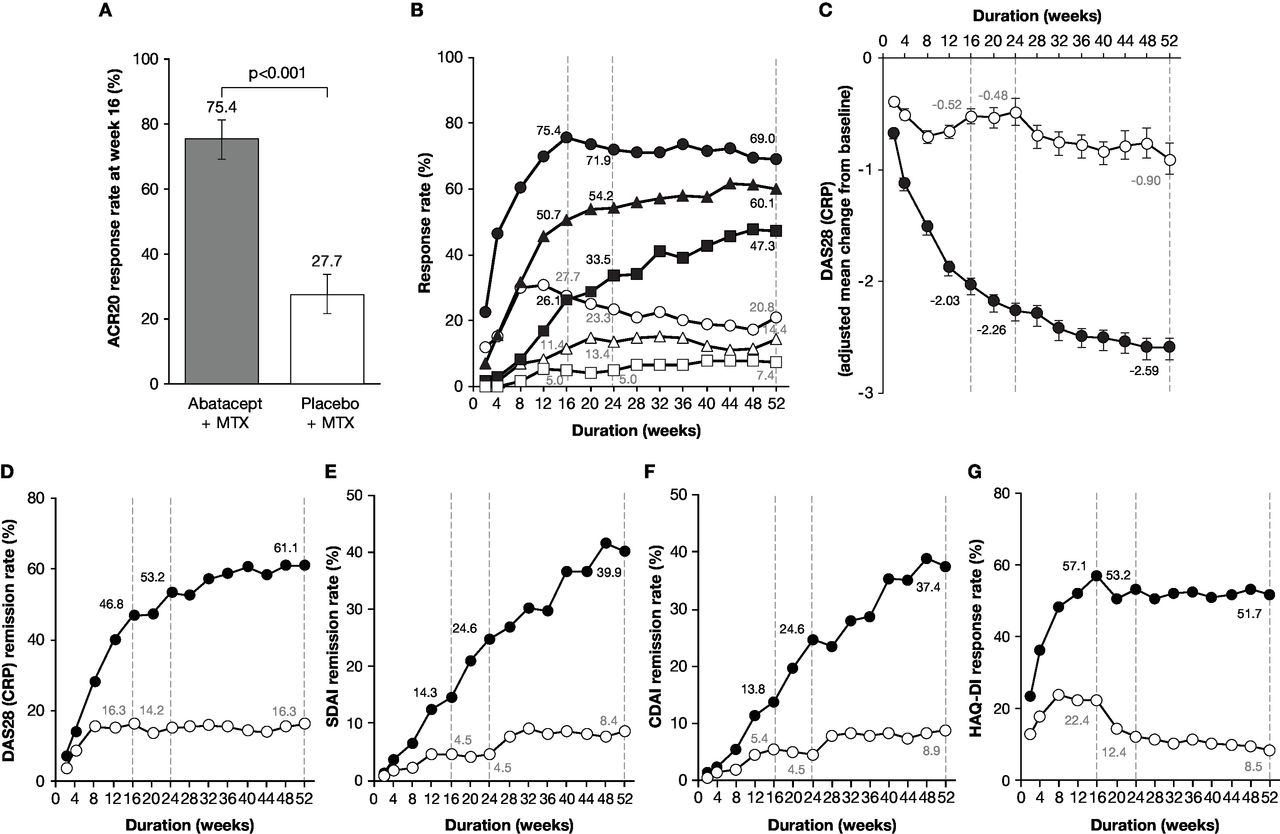
A significantly greater proportion of patients achieved ACR20 response at week 16 with abatacept plus MTX (153/203 patients (75.4%), 95% CI 69.4 to 81.3) compared with placebo plus MTX (56/202 patients (27.7%), 95% CI 21.6 to 33.9). The estimated difference between the groups was 47.6% (95% CI 38.6 to 56.7; p<0.001) (figure 2A). ACR20, 50 and 70 response rates over time from baseline to week 52 in the abatacept plus MTX and placebo plus MTX groups are shown in figure 2B. In the abatacept plus MTX group, ACR20 response was detectable at week 2, rapidly increased thereafter, and plateaued from week 16 to week 52. In contrast, ACR20 response in the placebo plus MTX group increased from week 2, reached a maximum at week 8 and gradually decreased thereafter up to week 52. Similarly, ACR50 and 70 responses in the abatacept plus MTX group improved rapidly from week 4 to week 8; were 50.7% and 26.1% at week 16 and 60.1% and 47.3% at week 52, respectively. Whereas, only marginal improvement was detectable for both parameters in the placebo plus MTX group. The adjusted mean change from baseline in DAS28 (CRP) rapidly decreased (week 4; greater than −1.0) with abatacept plus MTX, further improving thereafter (week 52; −2.59) (figure 2C), whereas it only decreased marginally with placebo plus MTX (week 52: −0.90). The score at week 16 was numerically greater in the abatacept plus MTX group versus placebo plus MTX (−2.03 vs –0.52, respectively; adjusted mean difference –1.50 (95% CI −1.73 to –1.28)). A higher DAS28 (CRP) remission rate (figure 2D) was also observed at week 16 with abatacept plus MTX (46.8%) compared with placebo plus MTX (16.3%) (estimated difference: 30.5, 95% CI 21.4 to 39.5), further increasing only with abatacept plus MTX up to week 52 (61.1% vs 16.3%; estimated difference: 44.7, 95% CI 35.8 to 53.7). SDAI remission rates (figure 2E) at weeks 16 and 52 with abatacept plus MTX (14.3% and 39.9%) were numerically higher than those with placebo plus MTX (4.5% and 8.4%). Similarly, CDAI remission rates (figure 2F) at weeks 16 and 52 were numerically higher with abatacept plus MTX (13.8% and 37.4%) than placebo plus MTX (5.4% and 8.9%). Changes in HAQ-DI responses for the abatacept plus MTX group were detectable from week 2 and rapidly improved thereafter, plateauing from week 16 (57.1%) to week 52 (51.7%) (figure 2G). In contrast, HAQ-DI responses in the placebo plus MTX group exhibited only marginal improvement within the first 16 weeks (22.4%) and gradually declined up to week 52 (8.5%).
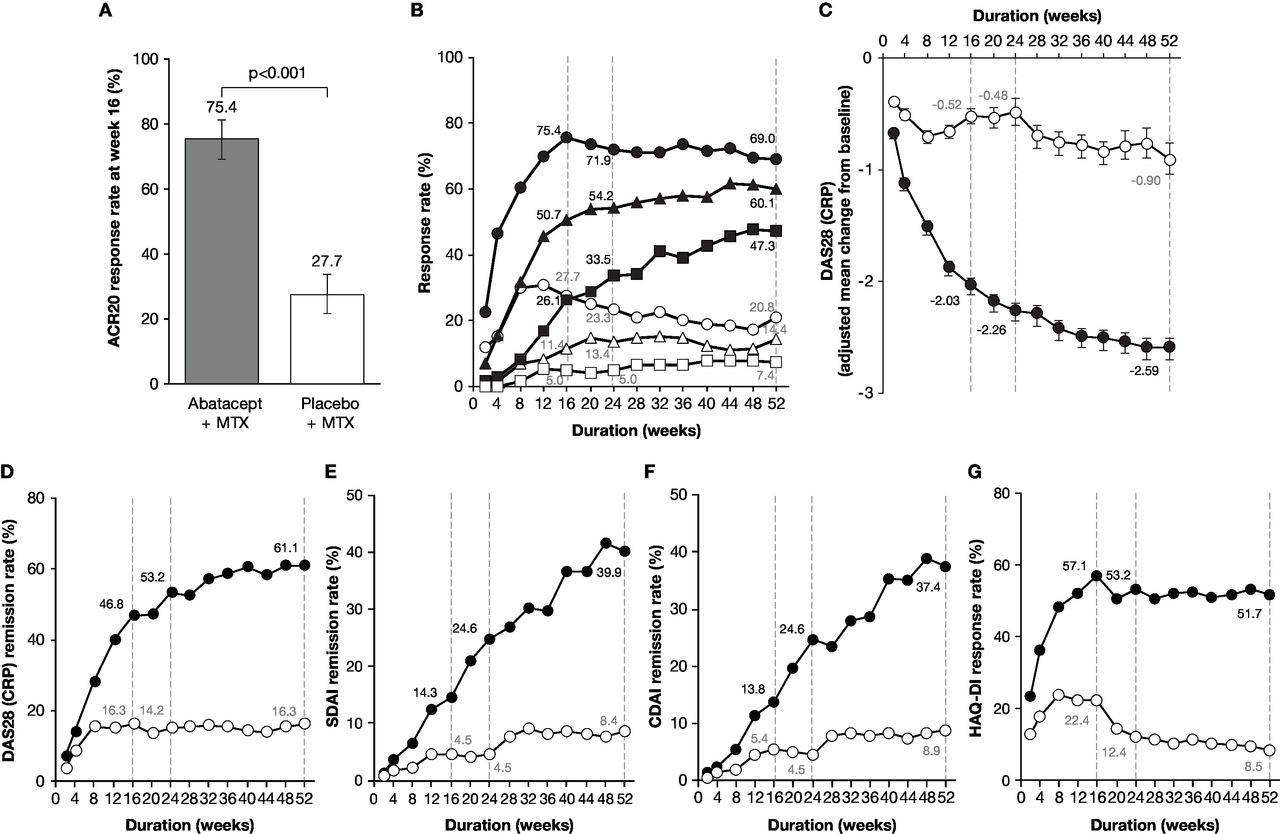
Disease activities evaluated as a comparison between abatacept plus MTX and placebo plus MTX groups. (A) ACR20 response rate (co-primary endpoint) assessed at week 16. P value was based on the continuity corrected χ2 test. (B) ACR20, 50 and 70 response rates over time up to 52 weeks. The patients who received rescue abatacept treatment, or who withdrew for any reason, were considered as non-responders from that time point forward. Dotted lines indicate time points at week 16 (secondary endpoints for ACR50 and 70 response rates), week 24 and week 52 (tertiary endpoints for ACR20, 50 and 70). The shapes indicate ACR20 (circles), ACR50 (triangles) and ACR70 (squares) response rates in the abatacept plus MTX (filled) and placebo plus MTX (open) groups. (C) Adjusted mean change from baseline in DAS28 (CRP) over time up to 52 weeks. Dotted lines indicate time points at week 16 (secondary endpoint), week 24 and week 52 (tertiary endpoints). Data after the initiation of rescue abatacept treatment were excluded. Error bars indicate the SE. (D–F) Remission rates with DAS28 (CRP) (D), SDAI (E) and CDAI (F) over time up to 52 weeks. Dotted lines indicate time points at week 16, week 24 and week 52 (tertiary endpoints). The circles show remission rates for the abatacept plus MTX (filled) and placebo plus MTX (open) groups. The remission was defined in this study as DAS28 (CRP) <2.6, SDAI ≤3.3 and CDAI ≤2.8. Patients with missing data due to early discontinuation or response after the initiation of rescue abatacept treatment were treated as non-responders. (G) Proportion of patients with HAQ-DI response over time up to 52 weeks. Dotted lines indicate time points at week 16, week 24 and week 52 (tertiary endpoints). The circles show response rates for the abatacept plus MTX (filled) and placebo plus MTX (open) groups. The HAQ-DI response was defined as a score reduction of ≥0.3 from baseline. ABA, abatacept; ACR20/50/70, American College of Rheumatology 20/50/70; CDAI, Clinical Disease Activity Index; CRP, C-reactive protein; DAS28, Disease Activity Score 28; HAQ-DI, Health Assessment Questionnaire-Disability Index; MTX, methotrexate; PBO, placebo; SDAI, Simplified Disease Activity Index.
Radiographic assessment
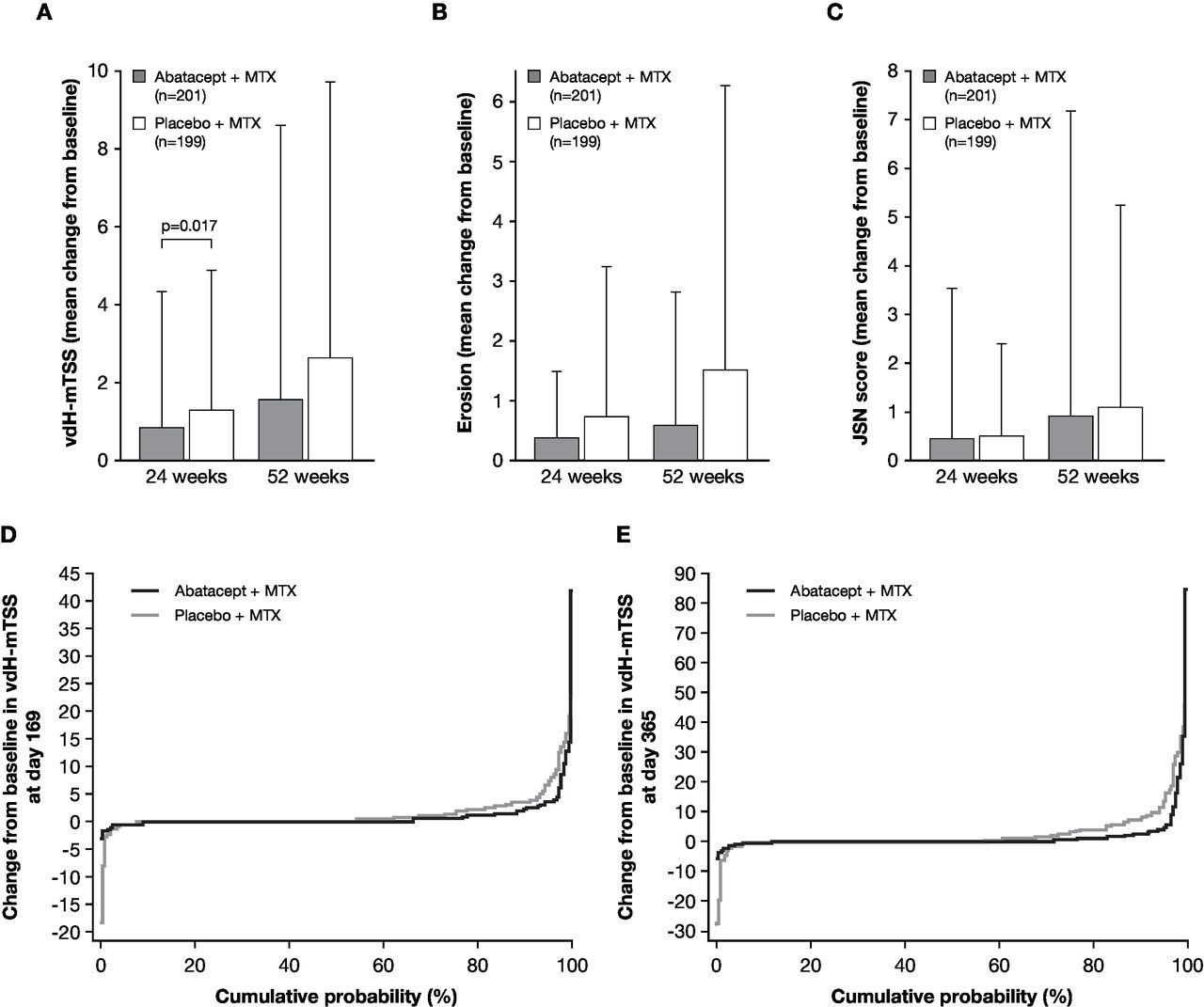
The mean change from baseline vdH-mTSS at week 24 was significantly lower with abatacept plus MTX than placebo plus MTX (0.84 vs 1.26; p=0.017; figure 3A). The mean change from baseline in vdH-mTSS at week 52 was numerically lower with abatacept plus MTX versus placebo plus MTX: 1.52 versus 2.62 (figure 3B). The numerical differences observed in mean change from baseline of erosion and JSN scores at weeks 24 and 52 are shown in figure 3B and C. Consistent with the vdH-mTSS, the mean change in erosion was numerically lower in the abatacept plus MTX group than in the placebo plus MTX group (week 24: 0.38 vs 0.74; week 52: 0.60 vs 1.52) (figure 3B). The change in JSN scores was relatively low in the abatacept plus MTX group compared with the placebo plus MTX group at week 24 (0.46 versus 0.52) and at week 52 (0.93 versus 1.10) (figure 3C). The sensitivity analyses demonstrated that mean (SD) change from baseline in vdH-mTSS at week 24 was 0.76 (2.86) in the abatacept plus MTX group and 1.20 (2.72) in the placebo plus MTX group by the analysis as observed, and 0.56 (1.53) in the abatacept plus MTX group and 1.00 (1.69) in the placebo plus MTX group by the analysis with multiple imputation.
{kind=link}
{kind=link}
{kind=link}
Joint damage progression evaluated as a comparison between abatacept plus MTX (grey bar) and placebo plus MTX groups (open bar). (A) The change from baseline in vdH-mTSS at week 24 (co-primary endpoint) and week 52. P value is based on a rank-based non-parametric analysis of covariance method. (B) The change from baseline in erosion score at week 24 and week 52 (tertiary endpoints). (C) The change from baseline in JSN score at week 24 and week 52 (tertiary endpoints). Data are shown as mean (SD). The number of patients with both baseline and post-baseline measurement are indicated in A–C. (D–E) Cumulative distribution function plot for the change from baseline in vdH-mTSS at week 24 (D) and week 52 (E). Missing data with patients who received rescue abatacept treatment, or who withdrew for any reason, were imputed with linear extrapolation. JSN, joint-space narrowing; MTX, methotrexate; vdH-mTSS, van der Heijde-modified total Sharp score.
The cumulative distribution function plots of change from baseline in vdH-mTSS of the abatacept plus MTX group and the placebo plus MTX group at weeks 24 and 52 are shown in figure 3D and E. The radiographic non-progressor rates in the abatacept plus MTX group and the placebo plus MTX group were evaluated by the proportion of patients with change from baseline in vdH-mTSS below SDC (1.9), 0.5 or 0 at week 24 and week 52 (see online supplementary table S1). The non-progressor rates below SDC in the abatacept plus MTX and the placebo plus MTX groups were 88.1% and 75.4% (estimated difference 12.7%, 95% CI 4.7 to 20.7) at week 24 and were 83.6% and 68.3% (estimated difference 15.2%, 95% CI 6.5 to 24.0) at week 52. Additionally, the non-progressor rates by the cut-off levels ≤0.5 or ≤0 at week 24 and week 52 were numerically higher in the abatacept plus MTX group than placebo plus MTX group.
Subgroup analysis
Subgroup analyses for ACR20 response rates at week 16 and for non-progression in change of mTSS at the cut-off level of ≤0 at week 24 are detailed in the online supplementary material and online supplementary figures S1A and B. Higher ACR20 response rates and non-progressor rates were observed in the abatacept plus MTX group in all subgroups examined (except RF-negative; details in the online supplementary material).
Safety
AEs and SAEs in the abatacept plus MTX and placebo plus MTX groups at weeks 16 and 52 are summarised in table 2. As a significant number of patients both in the abatacept plus MTX and placebo plus MTX groups received abatacept rescue therapy after week 16, the exposure-adjusted incidence rate (IR) per 100 person-years (PY) of exposure was calculated. Actual exposure of patients in the abatacept plus MTX and placebo plus MTX groups before entering abatacept rescue therapy up to week 52 were 212.9 and 108.7 PY, respectively. The exposure-adjusted IRs for SAEs throughout the study periods in the abatacept plus MTX and placebo plus MTX groups were 8.0 and 8.3 events/100 PY, respectively. In terms of AEs of special interests, the exposure-adjusted IR for infections in the abatacept plus MTX and placebo plus MTX groups were 78.4 and 95.7 events/100 PY, respectively. Notable clinical or laboratory test findings were not found in either group.
Summary of AEs reported in the 52-week study period
Discussion
To our knowledge, this is the first prospective, randomised, double-blind, placebo-controlled, multicentre study of abatacept in biologic-naive Japanese patients with RA who exhibited poor baseline prognostic factors. The co-primary endpoints demonstrated that abatacept in combination with MTX was superior to MTX alone, as evidenced by significantly higher ACR20 response rates at week 16 and lower mean change from baseline in vdH-mTSS at week 24. Furthermore, abatacept in combination with MTX was highly effective for the duration of the study, as compared with MTX alone, improving disease activity, ACR20/50/70 response rates, the mean change from baseline in DAS28 (CRP), remission rates (DAS28 (CRP)/SDAI/CDAI) as well as protection of joint damage destruction. Importantly, these positive clinical and radiographic outcomes with abatacept in combination with MTX were also consistently associated with improved patient-reported HAQ-DI response rates.
The achievement of clinical objectives for individual patients with RA, ‘treat-to-target strategy’ has been advocated by the EULAR recommendations2 21 and the ACR guidelines22 with adjustment/change of treatment if the target is not attained by 6 months. A few recent clinical studies for bDMARDs have focused on patients with ≤2 years of RA.23–31 In the present study, approximately 65% of patients had early RA with average disease duration of 1.8 years. Additionally, mean baseline mTSS levels, erosion and JSN were 11.0, 5.5 and 5.5, respectively, indicating the consistency of this study with the current clinical setting.32
The mean dose of MTX in this study was 9.6 mg/week, which is similar to the typical dose range used in Japan.33–35 In addition, recent clinical studies with a tumour necrosis factor-alpha (TNF-α) inhibitor have reported concomitant use of MTX at similar dose ranges.36 37 The present study reported similar ACR20, 50 and 70 response rates at week 24 with abatacept in combination with MTX to those for certolizumab and golimumab plus MTX studies.36 37
The impact of abatacept plus MTX treatment on joint damage progression, as observed by the mean change from baseline in vdH-mTSS at week 24 (0.84 vs 1.26 placebo plus MTX), differed from that reported for certolizumab plus MTX (0.2 vs 2.8 placebo plus MTX) and golimumab plus MTX (0.33 vs 2.51 placebo plus MTX).36 37 However, it should be noted that the mean change from baseline in vdH-mTSS in the placebo plus MTX group in the present study was lower compared with the other studies, implicating differences in patient populations. For example, the mean disease durations and vdH-mTSS among these studies differed by 1.7 years and 11.0 (present study), 5.8 years and 55.1 (Japan RA Prevention of Structural Damage (J-RAPID) study) and 8.7 years and 52.7 (GO-FORTH study), respectively, suggesting that the inhibition of joint destruction data should be carefully interpreted.36 37
In the subgroup analyses, abatacept plus MTX tended to be more efficacious than MTX alone for all subgroups examined, except in prevention of joint destruction in RF-negative patients. Wider estimated difference between abatacept plus MTX and placebo plus MTX in ACR20 achievement at week 16 was observed for patients with lower baseline DAS28 (CRP), mTSS or MTX dose. While differences in non-progression rates in mTSS at the cut-off level of ≤0 on week 24 tend to be shown for those with longer disease duration or RF positivity, the RF-negative subgroup also showed higher non-progressor rate than the RF-positive subgroup in both treatment arms.
In this study, the incidence of AEs and SAEs were similar in both treatment arms. Additionally, there were no significant increases in incidence of AEs and SAEs with abatacept plus MTX compared with placebo plus MTX over the dosing period. The safety profile of intravenous abatacept in this study was consistent with that observed in previous Japanese4 6 38 and international39–41 clinical trials.
The data presented here should be interpreted within the context of the study limitations.
In this study, abatacept improved disease activities and reduced progression of joint damage in ACPA-positive patients with RA who had moderate to severe disease activity and low radiographic joint damage.
Acknowledgments
Professional medical writing and editorial assistance was provided by Sharon Gladwin, PhD, and was funded by Bristol-Myers Squibb.
References
Footnotes
Contributors TM, KH, NA YT and TT contributed to study conception and design and interpreting data. TM, HI, TN, KT, AS, YS, KO, SN, YU, TH, YT and TT contributed to collecting data. KH performed the statistical analysis. NA managed the study. All authors contributed to critically reviewing and approving the manuscript.
Funding This study was sponsored by Bristol-Myers Squibb K.K. and Ono Pharmaceutical Co., Ltd.
Competing interests TM received speaker honoraria from Pfizer Japan, Nichi-Iko, Astellas, Meiji Seika, Bristol-Myers Squibb, AbbVie GK, Janssen, Chugai, Eisai, AYUMI. HI, AS, YS, KO, SN and YU have nothing to disclose. TN received grants from Pfizer Japan, AbbVie GK, Eisai, AYUMI. KT participated in speakers bureau for AbbVie, Astellas, Bristol-Myers Squibb, Eisai, Mitsubishi Tanabe. TH received grants from Eli Lilly, Bristol-Myers Squibb, AbbVie. KH and NA are employees of Bristol-Myers Squibb. YT received speaking and/or honoraria from Daiichi-Sankyo, Astellas, Eli Lilly, Chugai, Sanofi, AbbVie, YL Biologics, Bristol-Myers Squibb, GlaxoSmithKline, UCB, Mitsubishi Tanabe, Novartis, Eisai, Takeda, Janssen, Asahi Kasei and has received research grants from Mitsubishi Tanabe, Bristol-Myers Squibb, Eisai, Chugai, Takeda, AbbVie, Astellas, Daiichi-Sankyo, Ono, MSD, Taisho-Toyama. TT received grants from Astellas, Chugai, Daiichi-Sankyo, Takeda, AbbVie GK, Asahi Kasei, Mitsubishi Tanabe, Pfizer Japan, Eisai, AYUMI, Nippon Kayaku, Novartis K.K.; speaking fees from AbbVie GK, Bristol-Myers Squibb K.K., Chugai, Mitsubishi Tanabe, Pfizer Japan, Astellas, Daiichi-Sankyo, Eisai, Sanofi K.K., Teijin, Takeda, Novartis K.K. and consultant fees from AstraZeneca K.K., Eli Lilly Japan K.K., Novartis K.K., Mitsubishi Tanabe, AbbVie GK, Nippon Kayaku, Janssen K.K., Astellas, Taiho, Chugai, Taisho Toyama, GlaxoSmithKline K.K., UCB Japan.
Patient consent Not required.
Ethics approval The study was conducted in accordance with the Declaration of Helsinki,42 International Conference on Harmonisation Good Clinical Practice Guidelines,43 the Pharmaceutical Affairs Law, Good Clinical Practice and Good Post-marketing Study Practice as defined by the Ministerial Ordinance Concerning the Standards for the Implementation of Clinical Studies on Pharmaceutical Products and concerning notifications in Japan.44 The protocol, any amendments and the patient informed consent received local Institutional Review Board/Independent Ethics Committee approval prior to initiation of this study.
Provenance and peer review Not commissioned; externally peer reviewed.
Data statement Bristol-Myers Squibb policy on data sharing may be found at https://www.bms.com/researchers-and-partners/clinical-trials-and-research/disclosure-commitment.html.