Article Text

Abstract
Objective To describe the efficacy of tofacitinib in reducing pain in patients with rheumatoid arthritis (RA), psoriatic arthritis (PsA) or ankylosing spondylitis (AS) in a post-hoc analysis of randomised controlled trials.
Methods Data were collected from patients in seven tofacitinib studies: six phase III (four RA, two PsA) and one phase II study (AS), and grouped into five analysis populations based on rheumatic disease diagnosis and category of prior inadequate response (IR) to treatment: conventional synthetic disease-modifying antirheumatic drugs-IR (RA and PsA), tumour necrosis factor inhibitors-IR (RA and PsA), or non-steroidal anti-inflammatory drugs-IR (AS). Only patients who received tofacitinib 5 or 10 mg twice daily or placebo were included. Pain assessments included: Patient’s Assessment of Arthritis Pain, Short-Form Health Survey 36v2 Question (Q)7 and Bodily Pain domain, Ankylosing Spondylitis Quality of Life Q9 and Q14, EuroQol Five Dimensions Pain/Discomfort dimension and Bath Ankylosing Spondylitis Disease Activity Index Q2 and Q3. Data were reported to month 6 (placebo to month 3) in the RA and PsA populations, and week 12 (tofacitinib and placebo) in the AS population.
Results Overall, 3330 patients were included in this analysis. In the RA and PsA populations, pain improvements in tofacitinib-treated patients compared with placebo were observed at the earliest time point assessed and at month 3 (maintained to month 6). In the AS population, pain improvements compared with placebo were observed at week 12.
Conclusion Tofacitinib was associated with rapid and sustained improvements across multiple pain measures in patients with inflammatory rheumatic musculoskeletal diseases.
- rheumatoid arthritis
- psoriatic arthritis
- ankylosing spondylitis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Pain is the most common and impactful patient-reported symptom in inflammatory rheumatic musculoskeletal disease (RMD) and is considered important by both patients and healthcare professionals.
Tofacitinib is approved for the treatment of rheumatoid arthritis (RA), psoriatic arthritis (PsA) and moderate-to-severe ulcerative colitis, and is in development for the treatment of ankylosing spondylitis (AS).
What does this study add?
Across RA, PsA and AS, tofacitinib was associated with rapid or early alleviation of pain, and sustained improvements, assessed using both unidimensional pain measures and individual pain components of multidimensional measures.
Improvements appeared consistent, irrespective of tofacitinib dose or prior inadequate response to conventional synthetic disease-modifying antirheumatic drug, tumour necrosis factor inhibitor (RA or PsA) or non-steroidal anti‑inflammatory drug (AS) treatment.
How might this impact on clinical practice or future developments?
Tofacitinib was associated with rapid and sustained improvements across multiple pain measures in patients with inflammatory RMDs.
Introduction
Pain is the most common and most impactful patient-reported symptom in inflammatory rheumatic musculoskeletal diseases (RMDs).1 2 In a survey of 1204 patients with rheumatoid arthritis (RA), 68.6% reported pain as the most important area required for health improvement.2 The importance of pain in psoriatic arthritis (PsA) is demonstrated by its inclusion in a core domain set of disease features that should be measured in all clinical trials related to the treatment of patients with PsA.3 Similarly, in ankylosing spondylitis (AS), pain is included as part of a core set for monitoring patients with AS in clinical practice.4 Therapies that alleviate pain in inflammatory RMDs are therefore considered to be of high importance by patients and healthcare professionals.3
Traditionally, the pain experienced from RMDs was primarily attributed to peripheral nociceptive aetiologies (eg, inflammation or structural damage).5 However, patient reports of persistence of pain despite regression of inflammatory markers have highlighted the role of neurogenic mechanisms as significant factors in RMD-associated chronic pain.5–7 Chronic pain is a critical symptom of RMD progression, involving a multifaceted pathophysiological phenomenon including the release of various inflammatory factors, and peripheral and central pain-processing mechanisms (sensitisation).6 In recent years, the Janus kinase and signal transducer and activator of transcription (JAK-STAT) signalling pathway has been recognised as a key player in feedback loops involving pronociceptive and anti-inflammatory cytokines.8 Proinflammatory molecules may in turn sensitise neurons to pain signals. For example, patients with RA demonstrate enhanced sensitivity to nociceptive stimuli, and studies in rat models suggest that JAK/STAT signalling can promote mechanical pain sensitivity. Furthermore, studies in mice suggest that non-inflammatory molecules, such as the nociceptive chemokine CXCL1, may promote pain by activating sensory neurons.
Pain intensity and pain alleviation are important constructs that may be usefully evaluated in clinical trials.9 Patient-reported pain is typically measured using unidimensional questionnaires or single questions incorporated into a multidimensional assessment.10 Unidimensional measures may assess pain through a Visual Analogue Scale (VAS; from 0 mm (‘no pain’) to 100 mm (‘worst imaginable pain’)), or a Numeric Rating Scale for Pain (0 (‘no pain’) to 10 (‘worst imaginable pain’)). Such assessments are not specific to RMDs and can also be used in other patient populations. Generic multidimensional assessments applicable to various therapeutic areas and the general population (eg, Short-Form Health Survey 36v2 (SF-36v2) 11 and the EuroQol Five Dimensions questionnaire (EQ-5D)12 13), as well as those specific to RMDs (eg, American College of Rheumatology improvement criteria,14 Bath Ankylosing Spondylitis Disease Activity Index (BASDAI)15 and the Ankylosing Spondylitis Quality of Life (ASQoL) questionnaire16) also include pain as a key assessment within their frameworks.
Pain has been assessed in a large number of RA, PsA and AS randomised controlled trials (RCTs). In patients with RA, a network meta-analysis of 17 RCTs concluded that there was strong evidence that current biologic disease-modifying antirheumatic drug (bDMARD) interventions improve patient-reported pain, compared with placebo.17 This was also observed in studies in patients with PsA18–20 and a systematic review of studies in patients with AS,21 which reported that tumour necrosis factor inhibitors (TNFi) improved the symptoms of pain, compared with placebo.
Tofacitinib is an oral JAK inhibitor for the treatment of RA and PsA, and is under investigation for the treatment of AS. The efficacy of tofacitinib in improving patient-reported outcomes (PROs), including pain, has been demonstrated in PRO components of phase III RCTs in patients with RA22–26 and PsA,27 28 and in a phase II RCT in patients with AS.29
Pain reduction in patients treated with tofacitinib may be linked to the previously observed effect of tofacitinib on inflammation. For example, a statistically significant reduction in spinal inflammation has been reported in patients with AS receiving tofacitinib 5 mg and 10 mg twice daily (BID), compared with placebo.29 Furthermore, studies have reported that tofacitinib downregulates proinflammatory pathways in both RA and PsA models.30 31 Additionally, the suggestion that JAK/STAT signalling can promote mechanical pain sensitivity alludes to a possible link between tofacitinib and pain reduction,32 in addition to its proposed role in reducing inflammation.31
The objective of this post-hoc analysis was to use unidimensional pain measures and pain-specific components within multidimensional assessments to evaluate the efficacy of tofacitinib in reducing pain across three inflammatory RMDs (RA, PsA and AS) and subdivided according to inadequate response to previous therapies.
Patients and methods
Patients and study designs
This post-hoc analysis used data from the following double-blind, placebo-controlled RCTs, which all evaluated the impact of tofacitinib on patient-reported pain: four phase III RCTs in patients with RA (ORAL Scan (NCT00847613), ORAL Sync (NCT00856544), ORAL Standard (NCT00853385), ORAL Step (NCT00960440)); two phase III RCTs in patients with PsA (OPAL Broaden (NCT01877668), OPAL Beyond (NCT01882439)); and one phase II RCT in patients with AS (A3921119 (NCT01786668)).
The details of the individual trial designs and patient populations have been previously reported.29 33–38 In all studies, patients were aged ≥18 years and had a diagnosis of active disease at screening. Patients in the RA and PsA studies were required to be receiving concomitant conventional synthetic disease-modifying antirheumatic drug (csDMARD) treatment. Patients in the AS study were permitted to be receiving a stable dose of methotrexate (MTX) or sulfasalazine.
Patients with RA met the American College of Rheumatology 1987 Revised Criteria,33–35 39 and patients with PsA were classified based on fulfilment of ClASsification criteria for Psoriatic ARthritis.37 38 40 Studies were of 6–24 months’ duration in RA, and 6 or 12 months' duration in PsA, with patients in all studies randomised to receive tofacitinib 5 mg BID, tofacitinib 10 mg BID or placebo. In PsA studies (OPAL Broaden and OPAL Beyond) and ORAL Step (RA), patients treated with placebo were blindly switched to tofacitinib at month 3, or were blindly switched at month 3 if an insufficient therapeutic response was experienced. In ORAL Scan, ORAL Sync and ORAL Standard (RA), patients were blindly switched at month 6. In the ORAL Standard (RA) and OPAL Broaden (PsA) studies, patients were also randomised to an active control arm of adalimumab 40 mg once every 2 weeks (not included in this analysis).
Patients with AS fulfilled the modified New York criteria for AS (confirmed by centralised reading of sacroiliac radiographs),29 and had active disease based on a BASDAI score ≥4 and a back pain score (BASDAI Question (Q)2)≥4. All patients were randomised to receive tofacitinib 2 mg BID, tofacitinib 5 mg BID, tofacitinib 10 mg BID or placebo, which was maintained for the 12-week duration of the study. Patients randomised to tofacitinib 2 mg BID were not reported in this analysis so results could be compared with patients in the RA or PsA studies.
Patients from the seven RCTs were grouped into five analysis populations according to their rheumatic inflammatory disease (RA, PsA or AS) and inadequate response to previous therapies: patients with RA and inadequate response to csDMARDs (csDMARD-IR; pooled from three studies); patients with PsA and csDMARD-IR (one study); patients with RA and inadequate response to TNFi (TNFi-IR; one study); patients with PsA and TNFi-IR (one study); and patients with AS and an inadequate response to non-steroidal anti-inflammatory drugs (NSAID-IR; one study) (table 1).
Patient populations and studies
Pain assessments
Patient-reported pain was assessed using the following measures:
Patient’s Assessment of Arthritis Pain (PAAP) score (VAS 0–100 mm; higher scores indicated worse arthritis pain)41 was assessed in all RA and PsA populations at baseline, and months 1, 3 and 6. PAAP was also assessed at week 2 (ORAL Sync and ORAL Step (RA), and in both PsA populations), month 2 (ORAL Sync (RA), and in both PsA populations) month 4 (PsA populations), and month 4.5 (ORAL Sync and ORAL Step (RA)).
Patients achieving a decrease in PAAP score from baseline of ≥20 mm were assessed in all RA and PsA populations at months 1, 3 and 6. This was also assessed at week 2 (ORAL Sync and ORAL Step (RA), and in both PsA populations), month 2 (ORAL Sync (RA), and in both PsA populations), month 4 (PsA populations) and month 4.5 (ORAL Sync and ORAL Step (RA)).
SF-36v2 Q7: ‘How much bodily pain have you had during the past week?’ (range, 1–6 with 1=none, 6=very severe) and SF-36v2 Bodily Pain (BP) domain (norm-based score (theoretical range, 19.23–60.88),42 with higher scores indicating less pain) were assessed in RA populations at baseline, week 2 (ORAL Step only), and months 1, 3 and 6; in PsA populations at baseline and months 1, 3 and 6; and in the AS population at baseline and week 12.
EQ-5D Pain/Discomfort (PD) (range 1–3, with 1=no pain or discomfort, 3=extreme pain or discomfort)12 was assessed in RA and PsA populations at baseline, and months 1 (except ORAL Scan and ORAL Sync (RA)), 3, and 6, and in the AS population at baseline and week 12.
BASDAI Q2, assessing neck, back and hip pain (‘How would you describe the overall level of AS neck, back, or hip pain you have had?’) and Q3, assessing peripheral pain/swelling (‘How would you describe the overall level of pain/swelling in joints other than neck, back, hips you have had?’; VAS 0–10 cm in AS and VAS 0–100 mm in PsA; higher scores indicated worse pain), were assessed in the PsA populations at baseline and months 1, 3 and 6, and the AS population at baseline and weeks 2, 4, 8 and 12. In the PsA populations, BASDAI Q2 and Q3 were assessed only in patients who had spondylitis, as determined by the site investigator at screening, and baseline BASDAI total score >0.
Patients with a ‘yes’ response to ASQoL questionnaire Q9 (‘I have unbearable pain’) and Q14 (‘The pain is always there’) were assessed in the AS population at baseline and week 12.
Data analysis
Analyses were based on patients who received tofacitinib (5 mg or 10 mg BID) or placebo in one of the seven RCTs and had been included in the full analysis set (patients who were randomised and received ≥1 dose of study treatment).
Continuous endpoints (SF-36v2 Q7 and BASDAI Q2 and Q3) were summarised descriptively over time by mean and SE. Binary endpoints (PAAP response ≥20 mm, patients answering ‘yes’ to ASQoL Q9 and Q14) were summarised descriptively over time by number and percentage of patients (SE). For the RA and PsA populations, data from patients receiving tofacitinib were summarised up to 6 months, with placebo data summarised up to 3 months (last time point before placebo-treated patients switched to tofacitinib in some studies); for the AS population, all data were summarised up to 12 weeks.
In addition, for the RA and PsA populations, least squares (LS) mean changes from baseline for PAAP, SF-36v2 BP domain and EQ-5D PD dimension were analysed using a repeated-measures model with fixed effects for treatment group, visit, interaction of the treatment group by visit, geographic location, study (if more than one) and baseline value. The model used a common unstructured variance–covariance matrix, without imputation for missing data. Within this, two models were used: (1) up to month 3, the two placebo-to-tofacitinib sequences were combined into a single placebo group (pooled placebo group); (2) after month 3 (including all post-baseline data up to month 6), the placebo-to-tofacitinib sequences were kept separate. For the AS population, treatment comparisons between tofacitinib (5 mg or 10 mg BID) and placebo at week 12 were made using an analysis of covariance model, with fixed effects for treatment and baseline value; missing values were imputed by last observation carried forward.
Proportions of patients achieving a ≥20 mm decrease in PAAP score from baseline to month 3 were compared between tofacitinib (5 mg or 10 mg BID) and placebo, using large sample approximation of the difference in binomial proportions. When two or more studies were included, the Cochran-Mantel-Haenszel approach adjusting for study was used.
The cumulative distribution function plot for patients achieving specific changes from baseline in PAAP score was provided at month 3 in RA and PsA populations.43
P values were unadjusted.
Results
Patients
In total, 3330 patients were included in this post-hoc analysis. In each of the seven RCTs, baseline demographics/characteristics have been reported previously,29 33–36 44 45 and were generally similar in the tofacitinib and placebo treatment arms. Between the RCTs, patients in the RA population were generally older (mean age, 50.8–55.5 years) than those in the PsA (mean age, 46.9–51.3 years) and AS (mean age, 41.2–41.9 years) populations, with a longer average time since diagnosis (RA, 6.9–13.0 years; PsA, 5.3–9.6 years; AS, 1.5–4.1 years). Most of the RA population was female (80.2%–91.1%), compared with PsA, in which there were approximately equal proportions of male and female patients (47%–61% female), and the AS population, which was predominantly male (25.0%–37.3% female). In the RA and PsA analysis populations, csDMARDs were required for inclusion, with most patients receiving MTX. In the AS population, most patients (88.5%–94.1%) were receiving concomitant NSAIDs.
Between the TNFi-IR and csDMARD-IR populations in RA and PsA, baseline values for PAAP, SF-36v2 BP domain and EQ-5D PD dimension were generally similar.
Patient’s Assessment of Arthritis Pain
In the RA and PsA csDMARD-IR and TNFi-IR populations, significant improvements (reductions) in LS mean change from baseline in PAAP scores were reported as early as week 2 in the tofacitinib 5 and 10 mg BID treatment groups compared with placebo (figure 1). This significant improvement persisted for both tofacitinib treatment groups compared with placebo at month 3 (p<0.001; figure 1, table 2; pain scores for individual RA studies22 24 26 46 and for PsA have been reported previously27 28). The improvements from baseline in PAAP scores in the tofacitinib treatment arms were broadly maintained through month 6 (figure 1).

LS mean (SE) change from baseline in PAAP (measured using a VAS (0–100 mm; higher score=worse arthritis pain)) scores in (A) RA csDMARD-IR, (B) RA TNFi-IR, (C) PsA csDMARD-IR and (D) PsA TNFi-IR analysis populations (FAS). Significance is given as unadjusted p values, compared with placebo: *p≤0.05, **p<0.01, ***p<0.001. LS mean changes from baseline in PAAP were analysed using a repeated-measures model with fixed effects for treatment group, visit, interaction of the treatment group by visit, geographic location, study (if more than one) and baseline value. The model used a common unstructured variance–covariance matrix, without explicit imputation for missing data. Within this, two models were used: (1) up to month 3, the two placebo-to-tofacitinib sequences were combined into a single placebo group (pooled placebo group); (2) after month 3 (including all post-baseline data up to month 6), the placebo-to-tofacitinib sequences were kept separate. BID, twice daily; csDMARD, conventional synthetic disease-modifying antirheumatic drug; FAS, full analysis set; IR, inadequate response; LS, least squares; PAAP, Patient’s Assessment of Arthritis Pain; PsA, psoriatic arthritis; RA, rheumatoid arthritis; SE, standard error; TNFi, tumour necrosis factor inhibitor; VAS, visual analogue scale.
LS mean (SE) change from baseline in PAAP, SF-36v2 BP domain, and EQ-5D PD dimension to month 3*
In the same populations, the cumulative distribution function plots of patients achieving any level of decrease from baseline at month 3 in PAAP score were similar for patients receiving tofacitinib 5 mg or 10 mg BID, and greater than for patients treated with placebo (online supplementary figure 1). The proportions of tofacitinib-treated patients reporting a ≥20 mm improvement from baseline in PAAP scores at month 3 were significant across all these analysis populations compared with placebo (p<0.01; table 3). The proportion of tofacitinib-treated patients reporting improvements from baseline of ≥20 mm was maintained through month 6 (online supplementary figure 2).
Supplemental material
Percentage (SE) of patients reporting ≥20 mm improvements from baseline in PAAP at month 3*
Short-Form Health Survey 36 version 2
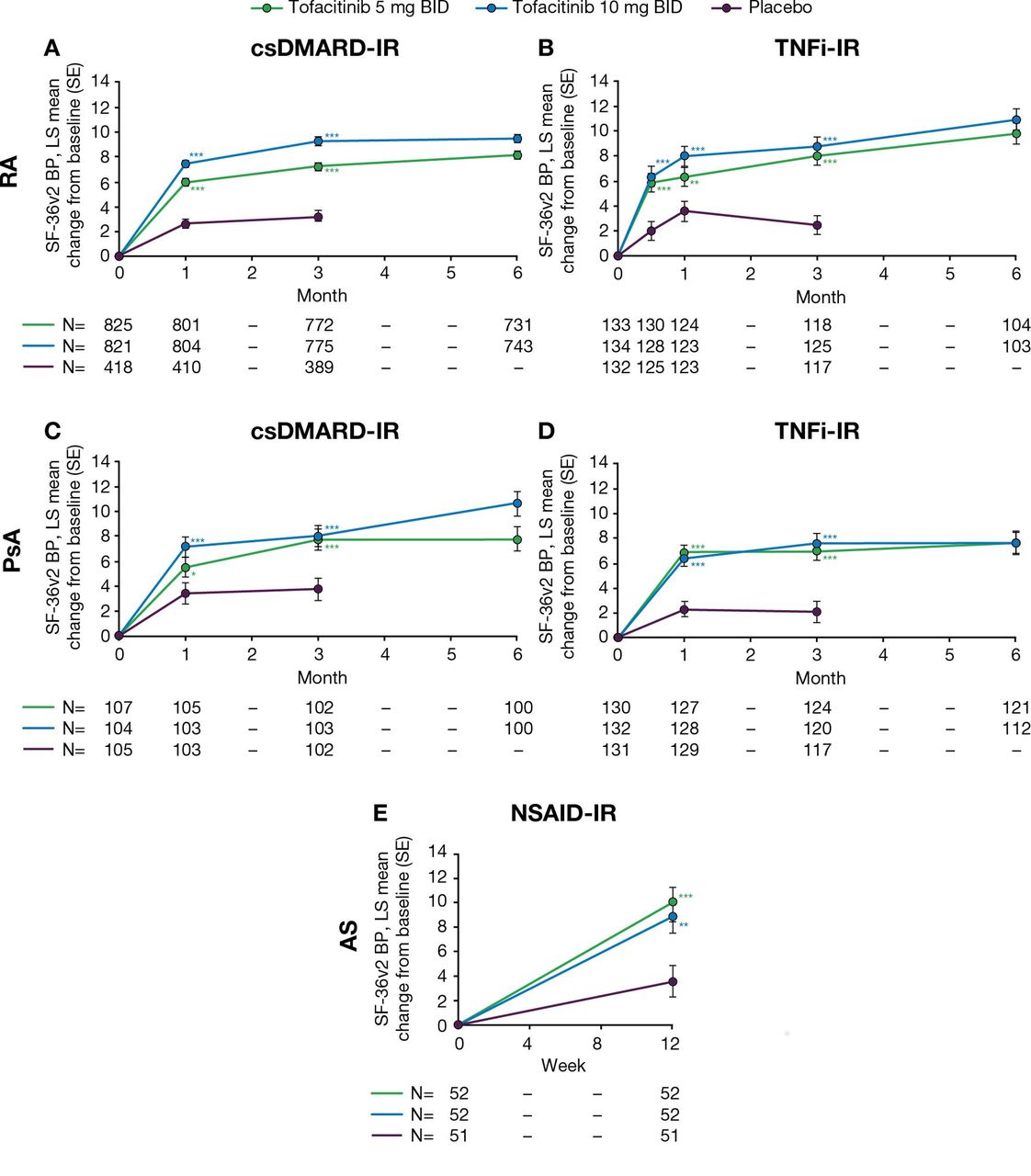
In the RA and PsA csDMARD-IR and TNFi-IR populations, improvements in mean SF-36v2 Q7 score (bodily pain during the past week; lower score indicated less pain; online supplementary figure 3) and LS mean change from baseline in SF-36v2 BP domain score (higher score indicated less pain; figure 2) were reported at the earliest available time point (week 2, RA TNFi-IR population; month 1, RA and PsA csDMARD-IR, and PsA TNFi-IR populations). Improvements in LS mean change from baseline in SF-36v2 BP domain for tofacitinib 5 and 10 mg BID treatment groups were significant at all time points compared with placebo (all p≤0.05; figure 2; table 2). In the tofacitinib 5 and 10 mg BID treatment groups, these improvements were maintained through month 6 (figure 2; online supplementary figure 3).

LS mean (SE) change from baseline in SF-36v2 BP domain scores (norm-based BP domain score (higher score=less pain)) in (A) RA csDMARD-IR, (B) RA TNFi-IR, (C) PsA csDMARD-IR, (D) PsA TNFi-IR and (E) AS NSAID-IR analysis populations (FAS). Significance is given as unadjusted p values, compared with placebo: *p≤0.05, **p<0.01, ***p<0.001. For RA and PsA populations, LS mean changes from baseline in SF-36v2 BP domain were analysed using a repeated-measures model with fixed effects for treatment group, visit, interaction of the treatment group by visit, geographic location, study (if more than one) and baseline value. The model used a common unstructured variance–covariance matrix, without imputation for missing data. Within this, two models were used: (1) up to month 3, the two placebo-to-tofacitinib sequences were combined into a single placebo group (pooled placebo group); (2) after month 3 (including all post-baseline data up to month 6), the placebo-to-tofacitinib sequences were kept separate. For the AS population, treatment comparisons between tofacitinib (5 mg or 10 mg BID) and placebo at week 12 were made using an analysis of the covariance model, with fixed effects for treatment and baseline value; missing values were imputed by last observation carried forward. AS, ankylosing spondylitis; BID, twice daily; BP, Bodily Pain; csDMARD, conventional synthetic disease-modifying antirheumatic drug; FAS, full analysis set; IR, inadequate response; LS, least squares; NSAID, non-steroidal anti-inflammatory drug; PsA, psoriatic arthritis; RA, rheumatoid arthritis; SE, standard error; SF-36v2, Short-Form Health Survey 36 version 2; TNFi, tumour necrosis factor inhibitor.
In the AS NSAID-IR analysis population, SF-36v2 Q7 (online supplementary figure 3), improvements were observed in patients treated with tofacitinib 5 mg or 10 mg BID compared with placebo at week 12 (the only post-baseline assessment). At week 12, LS mean change from baseline in SF-36v2 BP domain score was significantly greater with either dose of tofacitinib compared with placebo (both p<0.01; figure 2; table 2).
EuroQoL 5 Dimensions questionnaire Pain/Discomfort dimension
At month 3 (week 12 in the AS NSAID-IR analysis population), the LS mean changes in EQ-5D PD dimension scores were significant across both treatment groups and all analysis populations, compared with placebo (p≤0.05), with the exception of tofacitinib 5 mg BID in the AS NSAID-IR analysis population (p>0.05; table 2; online supplementary figure 4). These improvements in tofacitinib-treated patients were broadly maintained through month 6 (online supplementary figure 4).
Bath Ankylosing Spondylitis Disease Activity Index
In the PsA (only in patients who had spondylitis, as determined by the site investigator at screening, and baseline BASDAI total score >0) and AS populations, BASDAI Q2 (neck, back or hip pain) and Q3 (peripheral pain/swelling) scores were improved in both tofacitinib treatment groups and placebo compared with baseline, at month 3 and week 12, respectively (online supplementary figure 5). Improvements were numerically greater for patients receiving tofacitinib 5 or 10 mg BID compared with placebo. In PsA, improvements in these scores were generally maintained through month 6.
Ankylosing Spondylitis Quality of Life questionnaire
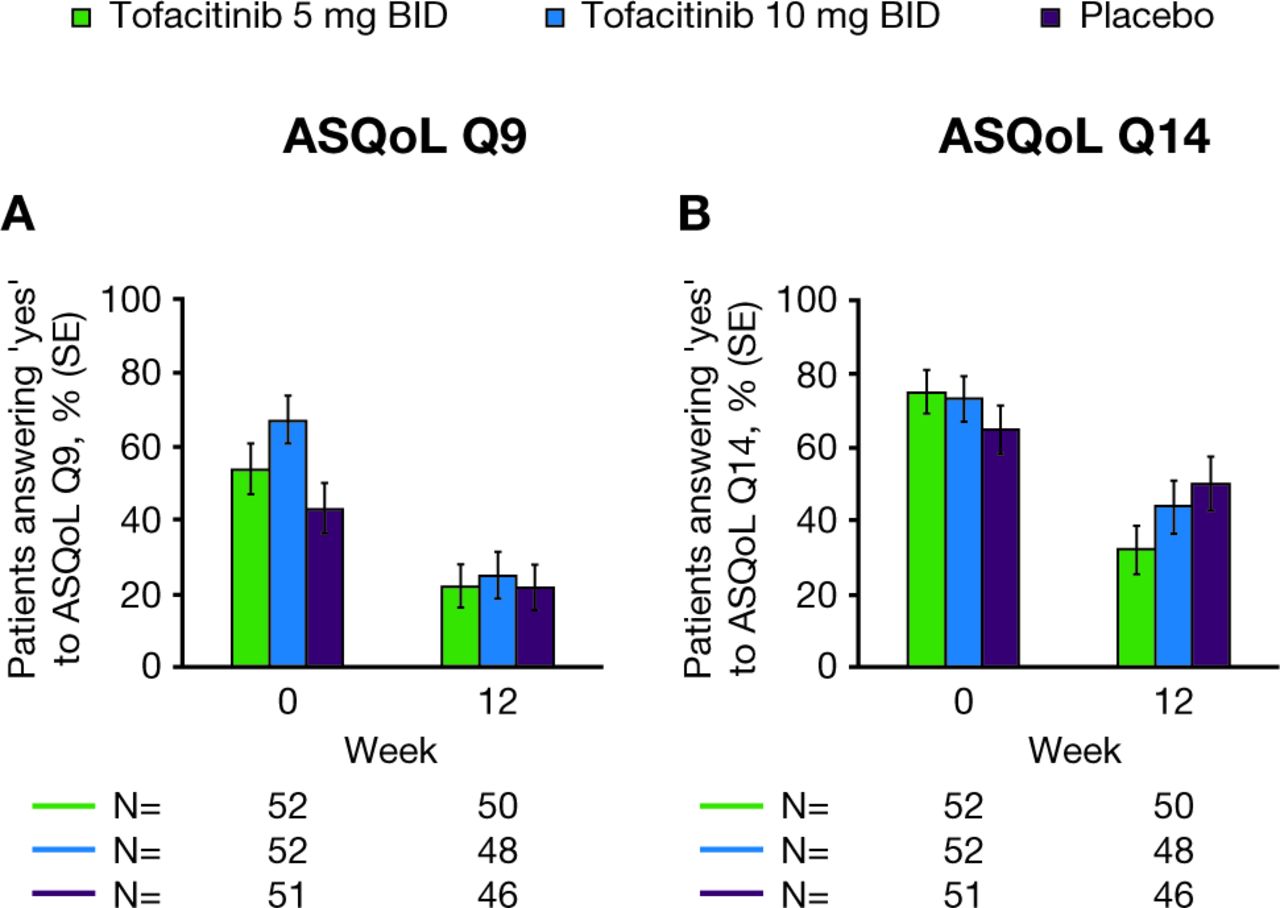
In the AS NSAID-IR analysis population, the percentages of patients who answered ‘yes’ to ASQoL Q9 or Q14 at week 12 were lower compared with baseline values in all treatment groups (figure 3). At week 12, the reductions from baseline in the percentages of patients who answered ‘yes’ to Q9 or Q14 were numerically greater in patients treated with tofacitinib 5 mg or 10 mg BID than those treated with placebo.

{kind=link}
{kind=link}
{kind=link}
Percentages (SE) of patients from AS NSAID-IR population answering ‘yes’ to (A) ASQoL Q9 and (B) ASQoL Q14. ASQoL Q9: ‘I have unbearable pain’; ASQoL Q14: ‘The pain is always there’. Missing values were not imputed. AS, ankylosing spondylitis; ASQoL Q9/14, Ankylosing Spondylitis Quality of Life questionnaire Question 9/14; BID, twice daily; IR, inadequate response; NSAID, non‑steroidal anti-inflammatory drug; SE, standard error.
Discussion
The objective of this post-hoc analysis was to further evaluate the efficacy of tofacitinib in reducing pain in patients with RA, PsA and AS treated in RCTs. While the effect of tofacitinib on pain has previously been reported in RA,22–26 PsA27 28 and AS,29 this analysis has considered relevant, pain-specific questions from multidimensional assessments, in addition to the unidimensional PAAP-VAS, across disease states and patient populations. This analysis has also evaluated all data in the context of three inflammatory RMDs and in patients exhibiting inadequate responses to previous therapies. Overall, patients in all populations treated with tofacitinib 5 mg or 10 mg BID showed improvements in all pain assessments, compared with placebo at month 3/week 12. As demonstrated in the ORAL Standard and OPAL Broaden studies, improvements in outcomes (including pain) were numerically similar between tofacitinib and active comparator (adalimumab) in the RA24 36 and PsA27 38 populations—these improvements were maintained in most outcomes through month 6. In the AS population, these improvements in patients receiving tofacitinib 5 mg or 10 mg BID also included the greater reduction from baseline in the percentages of patients answering ‘yes’ to ASQoL Q9 and Q14.
The experience of pain may be different between patients with RA, PsA or AS, due to the distinctive phenotypes and clinical pattern of presentation. For example, in PsA, the presence and severity of both skin and joint disease may change patients’ interpretation of pain relative to the potentially more joint-specific pain experienced by patients with RA. Despite this potential difference in pain experience, tofacitinib, regardless of dose, was associated with improvements across different aspects of pain across different disease types. Improvements were observed in general bodily pain (PAAP, SF-36v2 Q7 and BP domain, ASQoL Q9 and Q14, EQ-5D PD dimension), and in disease-specific neck, back and hip pain (BASDAI Q2).
Improvements were observed at the earliest measured time point and were similar across patients with an inadequate response to csDMARDs (RA and PsA), TNFi (RA and PsA) or NSAIDs (AS). This implies that tofacitinib efficacy in relieving pain has a rapid onset, and that efficacy may be similar between patients in more and less refractory populations. In patients with RA, analysis of time-to-event data for pain from ORAL Solo (NCT00814307; a phase III study comparing tofacitinib 5 mg and 10 mg BID monotherapy vs placebo), using an interactive voice-response system daily diary, demonstrated differentiation from baseline pain as early as 3 days after treatment initiation with tofacitinib.23 Furthermore, in the RA and PsA populations reported here, the proportions of patients reporting a ≥20 mm improvement were significantly higher for patients treated with tofacitinib compared with placebo. These results demonstrate that both tofacitinib doses (5 mg and 10 mg BID) are efficacious in the reduction of pain in many patients, regardless of inadequate responses to previous therapies.
The results of the current study appear consistent with previous results of bDMARD interventions in patients with RA, PsA or AS, with TNFi therapy shown to improve patient-reported pain in patients with RA (with an inadequate response to csDMARDs),17 PsA18–20 or AS.21 In patients with RA, other therapies such as abatacept, anakinra and the interleukin-6 inhibitor tocilizumab have also improved patient-reported pain.17 Furthermore, the magnitude of improvement in pain (as measured by VAS) with bDMARDs in RA and PsA appeared to show general agreement with the improvements in pain seen in patients who received tofacitinib, at similar time points (month 6/week 24).
One limitation of this post-hoc analysis is the difficulty in the measurement of pain, a multifactorial construct that incorporates both inflammatory and non-inflammatory concepts, with current pain measurements not definitively attributing pain to either cause. Specific to the AS population, it remains a challenge to confirm inflammatory back pain, as this is assessed solely based on patient input. Further factors limiting the comparison and interpretation of these results included the lack of confirmation of axial spondyloarthropathy in patients with PsA (determined by site investigator at screening rather than by imaging), the small AS population and differing time points. Additionally, the specific questions in the SF-36v2 (Q7), BASDAI (Q2 and Q3) and ASQoL (Q9 and Q14) analysed here were not developed to be presented as individual items, but rather to contribute to the total or domain-level scores and provide additional insight about which items are affecting those scores.15 16 42 Different time points used across study populations also limit the comparison and interpretation of the results. Finally, in this manuscript, we focused on the statistical significance of improvements in patient-reported pain within each disease state, rather than assessing similarities between levels of improvement across diseases. The varying designs of the different studies included in the analysis meant that a more formal meta-analysis to compare responses across diseases was not feasible or practical.
In summary, tofacitinib was associated with early and sustained improvements in pain, assessed using a range of measures, in patients with inflammatory RMDs. Improvements appeared consistent, irrespective of tofacitinib dose or prior inadequate response to csDMARD, TNFi (RA or PsA), or NSAID (AS) treatment. Further studies are needed to understand the mechanisms of pain in rheumatic diseases, how they differ between these diseases and how best to treat pain.
Acknowledgments
Medical writing support, under the guidance of the authors, was provided by Mark Bennett, PhD, of CMC Connect, a division of McCann Health Medical Communications Ltd, Glasgow, UK and was funded by Pfizer Inc, New York, NY, USA in accordance with Good Publication Practice (GPP3) guidelines (Ann Intern Med 2015;163:461-4).
References
Footnotes
Contributors AO, KdV, IBM, PJM, PB, TL, KK, CW, M-AH and AM conceived or designed the study. KK and CW analysed the data. KdV and PJM acquired data. All authors were involved in interpretation of data and reviewed and approved the manuscript’s content prior to submission. All author’s contributions to the current work met the relevant authorship criteria provided by the ICJME.
Funding This study was funded by Pfizer Inc.
Competing interests AO has served as a consultant for AbbVie, Amgen, BMS, Celgene, Corrona, Eli Lilly, Novartis, Pfizer Inc and Takeda, and has received grant support from Novartis and Pfizer Inc. KdV has received consulting fees or other remuneration from Eli Lilly and Pfizer Inc, and has participated in a trial with Galapagos. IBM has received research grants from Celgene, Janssen, Novartis, Pfizer Inc, Roche and UCB, and has received consulting fees or other remuneration from AbbVie, Celgene, Janssen, Novartis and UCB. PJM has received research grants from AbbVie, Amgen, BMS, Celgene, Eli Lilly, Janssen, Novartis, Pfizer Inc, Sun Pharmaceutical and UCB, has received consulting fees or other remuneration from AbbVie, Amgen, BMS, Celgene, Eli Lilly, Janssen, Novartis, Pfizer Inc, Sun Pharmaceutical and UCB, and is on the speakers’ bureau for AbbVie, Amgen, BMS, Celgene, Genentech, Janssen, Novartis, Pfizer Inc and UCB. PB has received consulting fees or other remuneration from AbbVie, Amgen, Eli Lilly, Johnson and Johnson, Novartis, Paladin, Sanofi-Genzyme and Takeda, and is on the speakers’ bureau for Amgen, Eli Lilly, Janssen and Pfizer Inc. TL, DG, KK, CW, and M-AH are employees and shareholders of Pfizer Inc. AM is a shareholder, and was an employee, of Pfizer Inc during the time of this analysis.
Patient consent for publication Not required.
Ethics approval All studies were carried out in compliance with the Helsinki Declaration and have received approval from the relevant Institutional Review Boards (IRB no.: 28306 [OPAL Broaden]; 28307 [OPAL Beyond]). Patients in all studies provided informed consent to participate.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request.