Article Text

Abstract
Background Tabalumab is a human monoclonal antibody that neutralises B-cell activating factor.
Objectives To evaluate tabalumab efficacy and safety in patients with rheumatoid arthritis (RA).
Methods This phase 3, randomised, double-blind, placebo-controlled study evaluated 456 patients with active RA after 24-week treatment with subcutaneous tabalumab (120 mg every 4 weeks (120/Q4W) or 90 mg every 2 weeks (90/Q2W)) versus placebo, with loading doses (240 or 180 mg) at week 0. Patients were allowed background disease-modifying antirheumatic drugs and previously discontinued ≥1 tumour necrosis factor α inhibitors for lack of efficacy/intolerance. Primary end point was American College of Rheumatology 20% (ACR20) response at 24 weeks. This study was terminated early due to futility.
Results Most patients had moderate-to-high baseline disease activity. There was no significant difference in week 24 ACR20 responses between 120/Q4W, 90/Q2W, and placebo (17.6%, 24.3%, 20%) per non-responder imputation analysis. Mean percent changes in CD20+ B-cell count (−10.8%, −9.6%, +10.9%) demonstrated expected pharmacodynamic effects. Treatment-emergent adverse events (AEs) were similar (59.5%, 51.7%, 52.6%), as were AE discontinuations (2.6%, 2.7%, 2.6%), serious AEs (4.6%, 4.1%, 3.9%), serious infectious events (1.3%, 0, 0) and events of interest: infections (23.5%, 25.9%, 24%), injection site reactions (13.1%, 25.8%, 11%) and allergy/hypersensitivity (3.9%, 4.1%, 3.9%) reports. Incidence of treatment-emergent antidrug antibodies was similar to placebo (3.9%, 4.8%, 3.9%). No deaths or new/unexpected safety findings were reported.
Conclusions Tabalumab did not demonstrate clinical efficacy in patients with RA in this phase 3 study, despite evidence of biological activity. There were no notable differences in safety parameters between tabalumab treatment groups and placebo.
Trial registration number: NCT01202773.
- Autoimmunity
- B cells
- Rheumatoid Arthritis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is already known about this subject?
B cells contribute to the immunopathology of autoimmune disorders including rheumatoid arthritis (RA), which may be related to increased B-cell activating factor (BAFF) signalling. Earlier phase clinical trials of anti-BAFF monoclonal antibodies showed a clinical effect in RA.
What does this study add?
BAFF targeting via tabalumab did not provide clinical benefit in this phase 3 trial for patients with moderate-to-severe RA with prior inadequate response to tumour necrosis factor (TNF) inhibitors.
How might this impact on clinical practice?
In patients with prior inadequate response to TNF inhibitors, targeting the BAFF pathway alone was not an effective approach to treating RA. Targeting BAFF may not be a viable therapeutic approach.
Introduction
B cells contribute to the immunopathology of autoimmune disorders including rheumatoid arthritis (RA), which may be related to increased B-cell activating factor (BAFF) signalling.1 Dysregulated BAFF expression contributes to autoimmunity primarily via effects on survival of immature and transitional B cells and the resulting failure to eliminate self-reactive B cells. Conversely, blocking BAFF has been shown to reverse autoimmune disease in animal models.2 ,3 Furthermore, many patients with RA have elevated BAFF in blood and synovial fluid.3 ,4
Disease-modifying antirheumatic drugs (DMARDs) are a part of the standard of care to treat RA, including the class of biologics (bDMARDs) that target tumour necrosis factor (TNF).5 Though numerous RA therapies are currently available, 20–50% of patients do not achieve significant clinical improvement,6–12 or they fail to maintain efficacy after initial therapeutic benefit.13 Thus, new treatment options for RA are needed.
Tabalumab is a fully human immunoglobulin G subclass 4 (IgG4) monoclonal antibody that binds and neutralises both soluble and membrane-bound BAFF.14 In phase 2 studies, tabalumab demonstrated evidence of both biological and clinical activity in patients with active RA and inadequate response to methotrexate.15 ,16 This phase 3 study was designed to evaluate efficacy and safety of tabalumab in patients with RA who had an inadequate response to one or more TNF inhibitors.
Methods
Study design
H9B-MC-BCDV (FLEX V; NCT01202773) was a phase 3, double-blind, placebo-controlled study comprised of a screening period of 7–28 days, a 24-week treatment period and post-treatment follow-up for up to 48 weeks. Participants were randomly assigned (1:1:1) to treatment groups by a computer generated random sequence using the Interactive Voice Response System (IVRS); the randomisation code was held by the vendor performing IVRS functions.
This study evaluated two subcutaneous (SQ) tabalumab doses: 120 mg every 4 weeks (120/Q4W) or 90 mg every 2 weeks (90/Q2W), versus placebo. At week 0, patients assigned to a tabalumab regimen received a SQ loading dose that was twice the treatment dose (ie, 240 mg or 180 mg).
Patient eligibility
Eligible patients were in American College of Rheumatology (ACR) functional class I, II, or III; had at least 8/68 tender and at least 8/66 swollen joints; had been treated at approved doses with at least 1 biological TNF inhibitor therapy; and stopped prior anti-TNF treatment due to either (1) insufficient efficacy or loss of efficacy after ≥90 days of treatment or (2) intolerance to treatment regardless of treatment duration. If patients were on conventional DMARDs, they were required to have been on a stable dose for ≥8 weeks prior to study baseline.
This study was conducted in accordance with local institutional review board ethical standards, good clinical practices and the Declaration of Helsinki. All patients provided written informed consent before study participation.
Study assessments
The primary objective was to demonstrate the superiority of either tabalumab regimen over placebo as measured by a 20% response rate in a core set of measures (ACR20) after 24 weeks of treatment.
Secondary efficacy end points were to demonstrate superiority of either tabalumab regimen over placebo after 24 weeks of treatment as measured by ACR50 and ACR70 (ie, 50% and 70% ACR response rates), ACR-N (per cent improvement on the ACR), individual components of the ACR core set, Disease Activity Score based on a 28-joint count (DAS28) and C reactive protein (CRP) level (DAS28-CRP), time to ACR20 response and European League Against Rheumatism Responder Index based on the 28-joint count (EULAR-28).
Health utilisation and outcomes evaluated as secondary end points included the Medical Outcomes Study 36-Item Short Form Health Survey (SF-36), Brief Fatigue Inventory (BFI), Brief Pain Inventory Modified Short Form (BPI-SF modified), duration of morning stiffness and the use of concomitant medications specifically for RA taken during the treatment period.
Biological activity of tabalumab was assessed over time via changes in serum immunoglobulins, CD20+ B-cell absolute counts and relative percentages (percentages of the total lymphocyte population), compared between each treatment regimen and placebo.
Safety assessments were treatment-emergent adverse events (TEAEs), TEAEs of special interest, clinical laboratory tests including immunogenicity testing, vital signs and concomitant medications.
Statistical analyses
A sample size of 555 randomised patients (185 patients each per tabalumab regimen and placebo group) was calculated to provide ≥99% power to detect a ≥30% difference in ACR20 response rates at week 24 for each tabalumab regimen versus placebo, assuming a placebo response rate of 18%. ACR20 significance testing used the Dunnett procedure at an overall 2-sided α level of 0.05, with each tabalumab regimen versus placebo comparison made at a two-sided α level of 0.0272. All other statistical tests of treatment effects and interaction effects were performed at two-sided significance levels of 0.05 and 0.10, respectively, unless otherwise stated. Primary and key secondary analyses followed a gatekeeping testing strategy to control the overall type I error rate at a two-sided α level of 0.05. Treatment group comparisons used Fisher's exact test for categorical data and analysis of variance (ANOVA) for continuous data, unless otherwise stated.
Efficacy and health outcome analyses were conducted following the intention-to-treat principle. Primary efficacy analysis was repeated on the per protocol population, a subset of the intent-to-treat (ITT) population excluding patients with significant protocol violations. Safety analyses were conducted on the safety population including all patients who received at least one dose of assigned study drug. Primary end point analyses of continuous efficacy and health outcome data were conducted using a modified baseline observation carried forward (mBOCF) approach; all other analyses were conducted using the modified last observation carried forward (mLOCF) approach. Non-responder imputation (NRI) was used for ACR analyses; non-responders (NR) were defined by <20% improvement in tender joint count and swollen joint count at week 16. Non-responders at week 16, patients who discontinued study treatment at any time and randomised patients with no postbaseline observations were defined as NR for all ACR end point analyses.
Results
Patient population
In total, 456 patients met enrolment criteria and were randomised, and comprised the ITT population: 153 patients in the 120/Q4W group, 148 patients in the 90/Q2W group and 155 patients in the placebo group (figure 1). Two randomised patients (1 patient each in the 90/Q2W and placebo groups) did not receive study treatment and were excluded from the safety population of 454 patients. The study was conducted from 28 January 2011 to 12 March 2013.
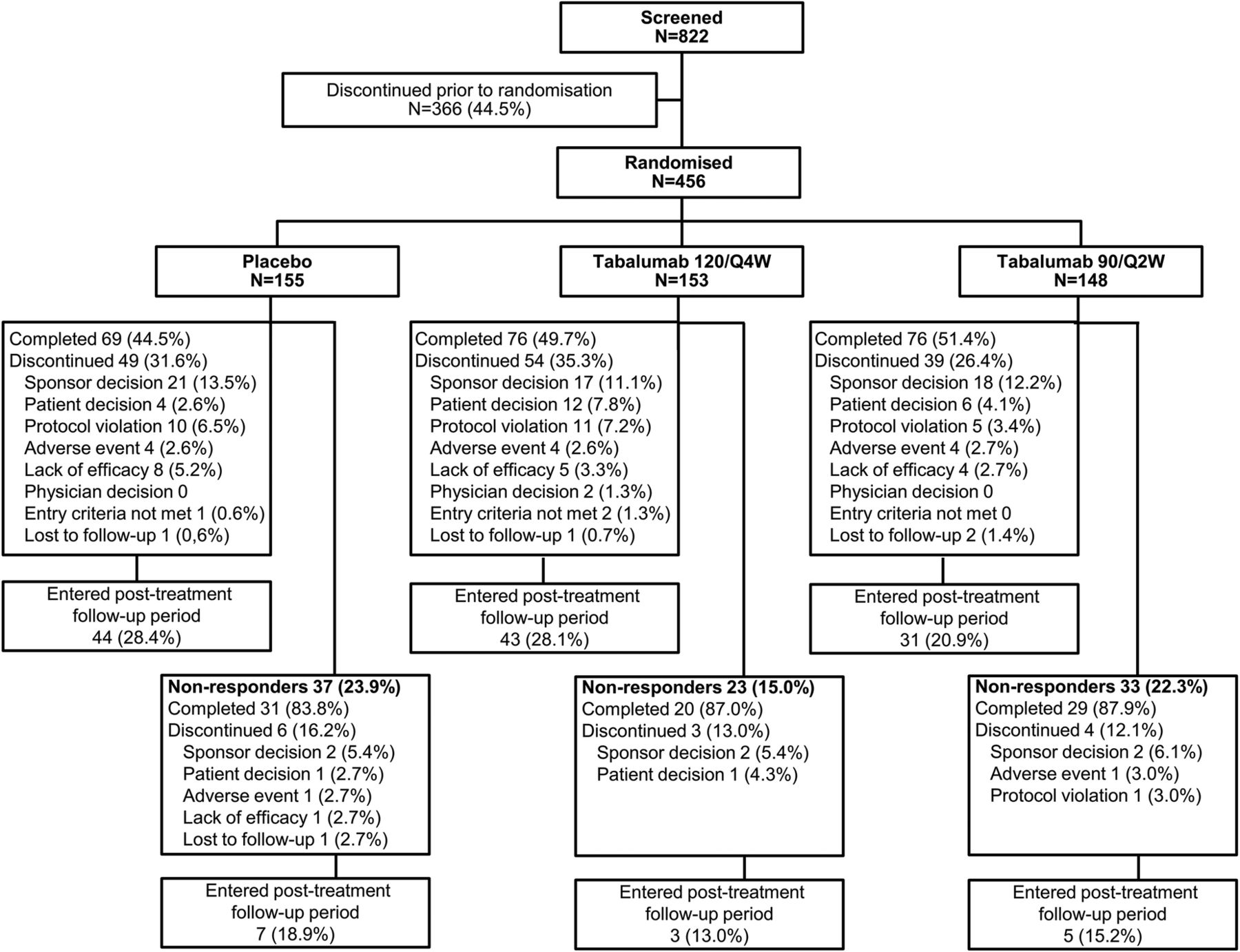
Patient disposition. Eligibility was assessed during screening, then randomisation to 24 weeks of treatment (or 16 weeks for non-responders) in 1 of 2 tabalumab regimens or placebo and 48 weeks of follow-up. 120/Q4W=120 mg subcutaneous (SQ) tabalumab injection every 4 weeks; 90/Q2W=90 mg SQ tabalumab injection every 2 weeks.
Baseline demographic and disease characteristics are summarised for each treatment group in table 1. Patients were a mean age of 53 years, the majority (84%) were women; most were located in North America (58.8%) with a mean time since RA diagnosis of 8.2 years. At baseline, most patients (75.4%) were seropositive for both RF+ and anti-CCP+. Patients for whom data were available had disease severity characterised as very active RA (75.3%), defined by DAS28-CRP >5.1, whereas the remaining patients had disease that was moderately active RA (24.7%), defined as DAS28-CRP >3.2–≤5.1. Demographic variables and clinical characteristics were generally balanced between treatment groups, with no significant difference between placebo and tabalumab treatment groups.
Patient baseline characteristics
Efficacy assessments
ACR20 responders at the week 24 end point in the ITT population included: 17.6% in the 120/Q4W group, 24.3% in the 90/Q2W group and 20% in the placebo group (figure 2). Fisher's exact test was used at the week 24 end point because the sample size was not sufficient for logistic regression due to week 16 non-response, drop out and sponsor decision. There were no significant differences in the ACR20 response rate at the week 24 end point for either tabalumab treatment group versus the placebo group; therefore, the primary end point of this study was not met. While a significant ACR20 response rate at week 12 was observed for patients in the 90/Q2W treatment group (28.4%) versus the placebo group (18.1%; p=0.030), this benefit was not sustained at week 24.

ACR20 response rates. ACR20 response rates based on non-responder imputation (NRI) for the 120/Q4W, 90/Q2W and placebo groups over 24 weeks of treatment. Response rates are based on the ITT population. ACR20=20% improvement in American College of Rheumatology criteria; 120/Q4W=120 mg subcutaneous (SQ) tabalumab injection every 4 weeks; 90/Q2W=90 mg SQ tabalumab injection every 2 weeks.
In the ITT population, mean changes from baseline at the week 24 mBOCF on individual ACR components—tender and swollen joint count, patient global assessment (PtGA), physician global assessment (PhGA), pain VAS—were similar across all treatment groups (data not shown). There were no significant differences for either tabalumab treatment group versus placebo in ACR50, ACR70, CRP, HAQ-DI or DAS28-CRP scores, or proportions of patients who reported a good to moderate rating on the EULAR-28 (mLOCF). After an interim analysis that was prompted by lack of efficacy in a separate tabalumab study (H9B-MC-BCDM; FLEX M; NCT01198002), the present study was terminated by the sponsor due to futility evidenced by insufficient efficacy.
Biological activity
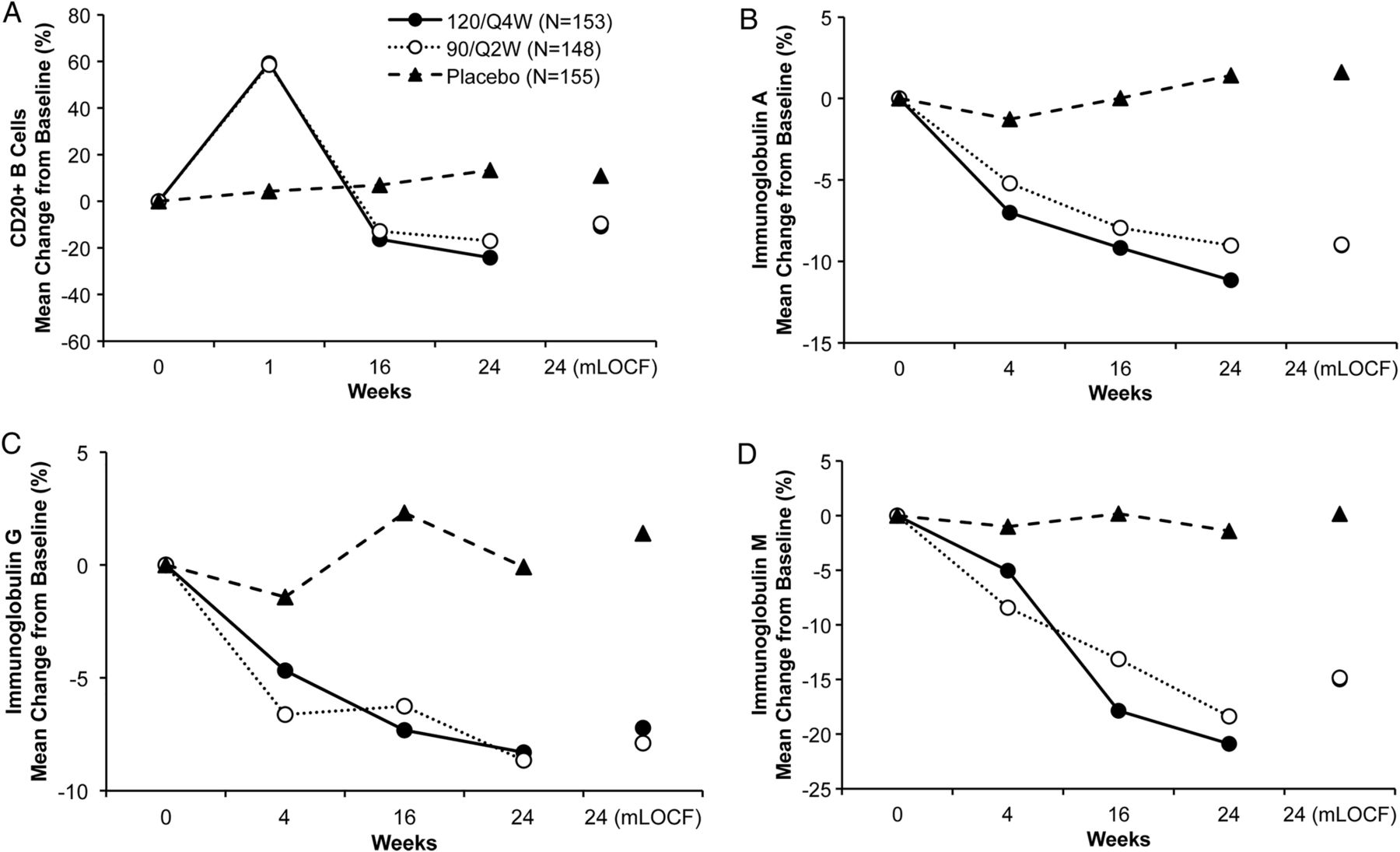
In the safety population, both tabalumab groups showed an expected initial increase in mean CD20+ B-cell absolute counts at week 1 (median per cent change from baseline: 37.6–56.3%) compared with placebo (−2.6%), followed by a subsequent decrease back to baseline or lower starting at week 4 (figure 3). At week 24 (excluding week 16 non-responders), CD20+ B-cell absolute count median per cent change from baseline was −43.2%, −53.2% and −1.1% for 120/Q4W, 90/Q2W and placebo groups, respectively. Significant differences were observed at week 24 (mLOCF) in tabalumab groups versus placebo for mean CD20+ B-cell count change from baseline (−62.0 cells/µL, −65.2 cells/µL, and −3.8 cells/µL; p<0.001 vs placebo for each comparison) and change from baseline in CD20+ B cells as percentage of total lymphocytes (−3%, −3.4%, and 0.1%; p<0.001 vs placebo for each comparison).

{kind=link}
{kind=link}
{kind=link}
(A–D). B-cell and Ig changes. Mean changes in total CD3-CD20+ B-cells (A) and immunoglobulin (Ig) subclasses levels (B-D) were measured over 24 weeks of treatment. Mean per cent changes are based on the safety population. 120/Q4W=120 mg subcutaneous (SQ) tabalumab injection every 4 weeks; 90/Q2W=90 mg SQ tabalumab injection every 2 weeks; mLOCF=modified last observation carried forward. *p<0.001 tabalumab groups versus placebo (both comparisons) at week 24 (mLOCF).
For week 16 non-responders initially randomised to tabalumab, the patterns of absolute CD20+ B-cell count median percent change from baseline and median time to B-cell nadir were similar with B-cell changes observed for the responders.
For this study, B-cell recovery was defined as <43 cells/µL and <50% of B-cell baseline values. Fifteen patients (excluding week 16 non-responders) had 1 or more low B-cell counts: 8, 5 and 2 patients, respectively. These patients were further evaluated to determine the time from nadir to recovery. The median time from B-cell nadir to recovery (Kaplan-Meier estimates, excluding week 16 non-responders) was 11.9 weeks (95% CI 8.4 weeks to not available) for 120/Q4W group and 12.3 weeks (95% CI 12.1 weeks to 22.4 weeks) for 90/Q2W group.
In the safety population, both tabalumab groups demonstrated decreases in mean serum immunoglobulins over the 24-week treatment period (figure 3B–D). At the week 24 end point (mLOCF), the IgA mean per cent change from baseline was −9%, −9% and +1.6%, respectively (p<0.001 vs placebo, each comparison); IgG mean per cent change from baseline was −7.2%, −7.9% and +1.4%, respectively (p<0.001 vs placebo, each comparison); and IgM mean percent change from baseline was −15%, −14.8% and +0.2%, respectively (p<0.001 vs placebo, each comparison).
No correlation was observed between immunoglobulin changes from baseline to the week 24 mLOCF end point and number of treatment-emergent infections or antidrug (tabalumab) antibodies (ADA) for either tabalumab group versus placebo during the treatment and follow-up periods.
Safety profile
During the 24-week treatment period, the incidence of treatment-emergent adverse events (TEAEs) and serious AEs were similar across treatment groups (table 2). The incidence of TEAEs was 59.5%, 51.7% and 52.6% in the 120/Q4W, 90/Q2W and placebo groups, respectively. The majority of TEAEs were mild or moderate in severity. The most frequently reported TEAEs (≥5% of patients in any group) were exacerbation of RA (5.9%, 4.8% and 7.8%) and upper respiratory tract infection (5.9%, 4.8% and 5.8%; table 2). There were no significant differences between either tabalumab group versus placebo for either of these events. Possibly-related TEAEs reported by ≥5% of patients in either tabalumab group included infections and infestations (7.2%, 9.5% and 11.7%), and general disorders and administration site conditions (7.2%, 9.5% and 5.8%). TEAEs were the reason for study discontinuation in four patients in each treatment group among responders. No significant dose-related increase in TEAEs was observed for any single event or grouping of TEAEs evaluated.
Summary of adverse events during the treatment period
AEs of special interest that deserve mention include infections, allergic/hypersensitivity events, injection site reactions, cardiovascular events and depression. The incidence of treatment-emergent infections (23.5%, 25.9% and 24%) and non-anaphylactic allergic/hypersensitive reactions (3.9%, 4.1% and 3.9%) were similar across 120/Q4W, 90/Q2W and placebo groups, respectively. Two major cardiovascular adverse events were reported during the treatment period: a serious arrhythmia (1 patient in the 90/Q2W group) and coronary revascularisation (1 patient in the placebo group). Twenty-one patients who received tabalumab (7 (13.1%) in the 120/Q4W group and 14 (25.8%) in the 90/Q2W group) and 6 (11%) patients in the placebo group reported a treatment-emergent injection site reaction during the treatment period. The exposure-adjusted rate of injection site reactions per 100 patient-years exposure for the 90/Q2W treatment group (25.8) was almost double the rate for the 120/Q4W (13.1) and placebo treatment groups (11.0). No patients discontinued study treatment due to an injection site reaction. All injection site reactions were mild to moderate in severity. The incidence of depression or suicidal ideation was similar across the treatment groups (2%, 0.7% and 2.6%).
Serious AEs were reported in seven patients (4.6%) in the 120/Q4W group, 6 (4.1%) in the 90/Q2W group and 6 (3.9%) in the placebo group, during the 24-week treatment period. Serious infections (2 cases of pneumonia, 1.3%) were reported in the 120/Q4W group only. Opportunistic infections were reported in 3 (2%), 6 (4.1%) and 6 (3.9%) patients, respectively, in the treatment and follow-up periods. Individual events included investigator reported pneumonia, nail infection, herpes zoster, upper respiratory infection, influenza, erysipelas, UTI, nasopharyngitis, sinusitis, cellulitis and device-related infection. No deaths were reported in this study.
No notable differences or trends were identified for vital signs, ECG, or clinical laboratory test results during the study treatment period.
Immunogenicity
Over the 24-week treatment period, 6 (3.9%), 7 (4.8%) and 6 (3.9%) patients in the 120/Q4W, 90/Q2W and placebo groups, respectively, developed persistent or transient treatment-emergent ADA (defined as ≥2 dilutions/fourfold increase from baseline). Four patients (2.6%), 2 (1.4%) and 4 (2.6%) patients, respectively, were classified as treatment-emergent ADA-persistent defined as 2 or more positive samples of at least 12 weeks duration.
Blood samples from patients who developed ADA were also evaluated for neutralising antidrug antibodies (NAb). One (0.3%) tabalumab-treated patient had a positive NAb result and 2 (0.7%) had negative results. Samples from 13 (4.3%) patients were inconclusive because drug concentrations exceeded the drug tolerance limit for the assay. Given the low frequency of ADA, it was difficult to assess the effect of ADA on the serum pharmacokinetics of tabalumab. No association was found between patients with treatment-emergent ADAs and reports of allergic/hypersensitivity (non-anaphylaxis) events or injection site reactions in the treatment or follow-up period.
Discussion
Prior phase two studies of tabalumab in patients with moderate-to-severe RA showed evidence of clinical efficacy, reporting higher ACR20 response rates and significant differences in response rates for ACR50, ACR70, DAS-28 CRP and EULAR scores in tabalumab groups versus placebo.15 ,16 Biological activity was also demonstrated in these earlier studies, including reductions in B-cell counts and serum immunoglobulins.
While there is no obvious explanation for the discrepant results in this study relative to the earlier phase two studies, there are some differences between the phase 2 and 3 studies to consider. The baseline characteristics for disease activity in the earlier phase 2 studies were generally comparable to the current study; however, this study was a global study, while some of the phase 2 studies enrolled patients from less diverse geographic regions and were smaller in size. In terms of study drug administration, in one phase 2 study of patients with inadequate response to TNF inhibitors, tabalumab was administered as either a 30 mg or 80 mg intravenous infusion at weeks 0, 3 and 6,17 while the current phase 3 study evaluated two subcutaneous (SQ) tabalumab doses of 120 mg every 4 weeks (120/Q4W) or 90 mg every 2 weeks (90/Q2W), versus placebo. A separate phase 2 tabalumab study of patients with inadequate response to methotrexate, which also applied intravenous administration and the same dosing frequency, demonstrated that all tabalumab doses were significantly more effective than placebo through week 24.15 It is notable that clinical efficacy corresponded to peak drug concentrations following infusion of tabalumab.
The current phase 3 study of patients with moderate-to-severe RA and prior treatment failure or intolerance to 1 or more TNF inhibitors did not meet its primary objective, as tabalumab treatment was not superior to placebo for the primary end point of ACR20 response at week 24 and did not meet any secondary efficacy objective. The efficacy of tabalumab in special patient subgroups (eg, prior prednisolone use, smoking status, RF/anti-CCP seropositivity status and number of previous TNF inhibitor treatment failures), was inconclusive because data were insufficient due to early termination of the trial. The findings in this study corroborate preliminary reported data from other BAFF inhibitor trials in patients with RA showing a lack of significant efficacy18 or efficacy that was restricted to a bDMARD-naïve subgroup.19 Despite the lack of efficacy, the biological activity of tabalumab was confirmed, but did not differ between the 2 dosing regimens in all parameters tested. Over the 24-week treatment period, CD20+ B-cells initially increased then gradually and significantly declined in response to tabalumab but not placebo administration, and without total B-cell depletion. This was expected, based on the mechanism of action of tabalumab: binding and neutralising BAFF,14 an essential B-cell survival signal, and has also been reported for other BAFF/B-lymphocyte stimulator (BLyS) inhibiting agents (belimumab, atacicept).20 Notably, these other BAFF blocking agents also showed a lack of correlation between different dosages and biomarker changes.21 ,22 In the current study, B-cell decline correlated with significant decreases in circulating IgA, IgG and IgM levels after tabalumab treatment. Despite decreases in biological parameters, there was no difference in infection rates for either tabalumab group compared with placebo. Tabalumab treatment was associated with a low incidence of ADA, and there was no association between treatment-emergent ADA and any reports of AEs.
Frequencies of TEAEs and SAEs were similar across tabalumab and placebo groups, and the incidence of AEs leading to study discontinuation was low across all treatment groups. The safety profile of tabalumab in this RA population was consistent with prior studies, and no new or unexpected safety findings were observed.15–17 Compared with prior phase 3 studies of other biological therapies in patients with RA, the present study of tabalumab had a similar incidence of TEAEs (range: 51.7-59.5%), discontinuations due to TEAEs (range: 2.6–2.7%), infections (range: 23.5–25.9%) and SAEs (range: 3.9–4.6%).6 ,23–26
There were limitations to the design and interpretation of results of this study, which was terminated early. The effects of background DMARD treatment, and prior and concomitant RA medications, may limit interpretation of results. In addition, patients were previously treated with 1 or more TNF inhibitors and had discontinued due to treatment failure or intolerance; there may be patients in this population who are refractory to immunomodulatory biological therapy for RA, including tabalumab.
In this phase 3 study in patients with previous failure of biological therapy, tabalumab did not demonstrate clinical efficacy in either dosing regimen, despite evidence of biological activity. Targeting BAFF may not be a viable therapeutic approach. Alternatively, the dose of active study drug and/or duration of the study may not have been optimal to observe a clinical effect. No new or unexpected safety findings were reported for patients with RA who received tabalumab.
Acknowledgments
The authors thank Jennifer N Bodie, PhD and Teresa Tartaglione, PharmD (ClinGenuity, LLC, Cincinnati, OH), for medical writing support and assistance with preparation and submission of this paper. This study was sponsored by Eli Lilly and Company.
References
Footnotes
Contributors MS, BC, TD, JMK and TWH were study investigators, and participated in the interpretation of data and data reporting. MS, AG, WK, P-YB, RO and CL contributed to conception of the study design and analysis plan, data interpretation and data reporting. All the authors provided critical input and approval to the final manuscript.
Funding This study was sponsored by Eli Lilly and Company. The protocol for this trial may be found at: https://clinicaltrials.gov/ct2/show/NCT01202773?term=NCT01202773&rank=1.
Competing interests MS reports consultancy fees with AbbVie, Amgen, Antares, Bristol-Myers Squibb, Eli Lilly, Horizon, Johnson & Johnson, Novartis, Novo Nordisk, Pfizer, Roche and UCB Pharma; is a member of AbbVie's speaker bureau; and has received research funding from UCB Pharma. BC reports personal fees and other from Eli Lilly during the conduct of the study; grants and personal fees from Bristol Myers Squibb; personal fees from Janssen; grants and personal fees from Merck; grants and personal fees from Pfizer; grants and personal fees from Roche-Chugai; and grants from UCB Pharma. TD reports grants and personal fees from UCB Pharma; grants and personal fees from Roche/Chugai, grants and personal fees from Sanofi; and grants from Janssen and Johnson & Johnson. JMK reports fees for research and consulting from Eli Lilly. TWH reports fees for research and for serving on the scientific advisory board from Eli Lilly. MV, AG, WK, P-YB, RO and CL are all employees and stockholders of Eli Lilly. WK also reports owning stocks and receiving dividends from the Pfizer Corporation.
Ethics approval Institutional Review Board at each investigative site.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.