Article Text

Abstract
The recent identification of genetic mutations leading to dysfunction of inflammatory and apoptotic pathways, has allowed to characterise a group of diseases, recognised as monogenic autoinflammatory syndromes. Among those, Blau syndrome (BS) and early-onset sarcoidosis (EOS) have been identified as familial and sporadic phenotypes of the same non-caseating granulomatous form. Both the diseases are caused by mutations in the CARD15/NOD2 gene, encoding the cytosolic NOD2 protein, one of the key molecules in the regulation of innate immunity. Clinical onset is typically located in the first years of life and phenotype is characterised by simultaneous or less articular, cutaneous and ocular non-caseating granulomatous inflammation, which can be variably associated with a heterogeneous systemic spectrum. The CARD15/NOD2 gene has also been identified as one of the genes linked to susceptibility to Crohn's disease (CD), a common polygenic inflammatory granulomatous bowel disease. The heightened nuclear factor-κB activity, found in the intestinal tissue of patients affected by CD, has probably a genetic cause related to several CARD15/NOD2 polymorphisms. Other substitutions in the CARD15/NOD2 gene have also been found in a recently described disorder, called NOD2-associated autoinflammatory disease, which shares several clinical characteristics with BS and EOS. This review attempts to describe these diseases on the basis of the most recent evidences. We described genetic and clinical aspects, mainly focusing on BS and EOS, the most representative diseases of autoinflammatory granulomatous diseases, with the ultimate purpose to expand their knowledge.
- Fever Syndromes
- Inflammation
- Arthritis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
Blau syndrome (BS) and early-onset sarcoidosis (EOS) caused by mutations in the CARD15/NOD2 gene, represent familial and sporadic phenotypes of the same non-caseating granulomatous inflammatory disease.
The CARD15/NOD2 gene has also been identified as one of the genes linked to susceptibility to Crohn's disease (CD).
Other substitutions in the CARD15/NOD2 gene have also been found in a recently described disorder, called NOD2-associated autoinflammatory disease (NAID).
The improvement of knowledge in molecular mechanisms and diagnosis of these diseases can represent an important breakthrough for improving their outcomes.
Introduction
In past years, the recognition of different mutations in genes involved in the regulation of inflammatory and apoptotic cellular mechanisms has allowed the characterisation of monogenic autoinflammatory syndromes.1
Despite their genetic and clinical heterogeneity, the common starting mechanism of these diseases has been identified in the dysfunction of innate immunity and inflammasome processes.1 Physiologically, the inflammasome represents a multiprotein cytoplasmatic platform that, when assembled, following a proinflammatory trigger, activates caspases, which convert prointerleukin (IL)-1β into its active form, IL-1β, a key molecule of inflammation and innate immunity.2
In monogenic autoinflammatory syndromes, a genetic-driven erroneous assembly of the inflammasome leads mainly to an overstated increase in IL-1β with an aberrant inflammatory response.2 Recurrence of the inflammatory attacks are the main temporal characteristics of these diseases, thus reflecting their spontaneous and unbalanced nature.1 ,2 Although the knowledge of IL-1β and inflammasome mechanisms represents the major conceptual progress in the autoinflammatory processes, the recognition of various genetic backgrounds has been useful to classify different groups of autoinflammatory disorders.1 Among these diseases, two rare non-caseating granulomatosis, Blau syndrome (BS) and early-onset sarcoidosis (EOS), caused by sequence variants in CARD15/NOD2 gene, have been included in the group of nuclear factor κB (NF-κB) activation disorders or autoinflammatory granulomatous diseases.1 ,3
Further, studies focusing on genetic background in Crohn's disease (CD) have highlighted its susceptibility in patients carrying several CARD15/NOD2 polymorphisms.4 Most recently, a positive association with other CARD15/NOD2 gene polymorphisms has been recently described in a systemic syndrome, termed NOD2-associated autoinflammatory disease (NAID).5
The purpose of this review is to describe BS and EOS, with the main aim of improving knowledge of genetic and clinical aspects.
We provide a brief description of NAID and we review main pathogenetic and clinical aspects behind a positioning of CD, in autoinflammatory granulomatous diseases.
Blau syndrome and early-onset sarcoidosis
Genetic and pathogenic aspects
In 1985, Dr Blau and Dr Jabs first described a dominant inherited form of granulomatosis with paediatric onset, which is also recognised as “Blau syndrome” and “Jabs syndrome”.6 ,7 The clinical picture was represented by recurrent non-caseating granulomatous symmetric arthritis, dermatitis, uveitis and cranial neuropathies.6 ,7 BS and in EOS,8 showing clinical similarities, were grouped in the set of juvenile granulomatous systemic disorders or paediatric granulomatous arthritis.8 Successively, both the diseases were established as the familial and sporadic forms of the same non-caseating granulomatous autoinflammatory spectrum.9 ,10 Indeed, BS and EOS, in addition to sharing the same clinical manifestations,11 ,12 showed the same genetic background, represented by mutations of the CARD15 (caspase activation and recruitment domain (CARD) family, member 15)/NOD2 (nucleotide-binding oligomerisation domain containing 2) gene.13 ,14
CARD15/NOD2 is mapped on chromosome 16q12 and encodes the multidomain cytosolic protein of almost 1000 amino acids, namely nucleotide-binding oligomerisation domain containing 2 (NOD2).15–17 NOD2 is a member of a family of pattern recognition molecules, with N-terminal CARDs, also called the nucleotide-binding domain and the leucine rich repeat (LRR)-containing family (Nod-like receptors, NLRs).15–17 NOD2 is primarily expressed in the cytosol of the antigen-presenting cells as myelomonocytes (monocytes/macrophages and granulocytes), dendritic cells, intestinal Paneth cells and the plasmatic membranes of gut epithelial cells.16
The three-domains structure of the protein contains two N-terminal CARDs for downstream signalling through CARD–CARD interactions, a central nucleotide-binding and oligomerisation domain (NACHT) with ATPase activity, and nine C-terminal LRRs that sense pathogen-associated molecular patterns, such as muramyl dipeptide and various degradation bacterial products.16
CARD15/NOD2 mutations described in BS and EOS are mainly located in the NACHT domain and while decreasing the spontaneous oligomerisation process of the mutated protein, they hyperactivate the NF-κB responsive reporter gene and increase basal NF-κB activity, even in the absence of stimulation.17
In particular, NOD2 is physiologically folded in an inactive structure by the binding of NACHT to the LRR domain.17 Bacterial stimulation induces a NOD2 conformational change and protein activation by exposure of the CARD domains and recruitment of the serine/threonine kinase receptor-interacting protein kinase 2 (RIP2), through a CARD–CARD interaction.17 RIP2 consequently promotes the activation of NF-κB, mitogen-activated protein kinase (MAPK), and the interferon regulatory factor family.17
RIP2 is able to induce the NF-κB essential modulator ubiquitination, and consequently Iκ-kinase (IKK) complex activation, which allow the phosphorylation and degradation of the NF-κB inhibitor IκBα.17 Thus, activated NF-κB can translocate to the nucleus, with activation of proinflammatory transcriptional genes.17 Additionally, RIP2 can activate MAPK by recruitment and activation of transforming growth factor β-activated kinase 1 (TAK1), a crucial regulator in cellular differentiation and proliferation.15–17 Moreover, in response to viral single-stranded RNA involvement, NOD2 interacts with the mitochondrial antiviral-signalling protein and thus induces NF-κB and tumour necrosis factor receptor-associated factor 3 (TRAF3) signalling pathways activation.17
The effects of CARD15/NOD2 mutations in BS and EOS are not fully understood. Although evidence continues to be poor, a possible role of mycobacterial components has been suggested as triggers of granulomatous autoinflammation.18 ,19
Improvement in the knowledge of pathogenic aspects of BS and EOS started with the identification of three missense mutations (p.R334Q, p.R334W and p.L469F) in four European families with BS.13 To date, on a total amount of almost 220 patients with BS and EOS carrying CARD15/NOD2 mutations, missense substitutions involving residue at position 334, p.R334Q/W, account for more than 80%.3 ,20–22 p.E383K has been found in almost 5% of cases, whereas other mutations have been described most rarely. Notably, in two patients with EOS, the concomitant presence of p.E338D and p.D390V mutations23 and a NOD2 six-base deletion (c.1493_1498delAACTGT, p.E498V, 499-500del) has been described.24 A schematic representation of the NOD2 protein and BS and EOS-related mutations are described in figure 1.

Schematic representation of the NOD2 protein (NP_071445.1) with the characteristic three domains structure and localisation of NOD2 mutations described in autoinflammatory granulomatous diseases. Red squares highlight mutations associated with BS, whereas the grey colour identifies EOS-related mutations, on the basis of the Infevers database data (CARD, caspase recruitment domain’ LRR, leucine rich repeat; NACHT, central nucleotide-binding and oligomerisation domain; http://fmf.igh.cnrs.fr/ISSAID/infevers/index.php).
Clinical aspects: the classic triad
Articular findings
Data on clinical aspects of BS and EOS are mainly derived from case reports or case series,12 international registries,20–22 and more recently by a 3-year multicentric observational study.11 The prevalence and incidence of BS and EOS still remain unknown.3 Onset occurs in paediatric age and usually under 5 years of age.11
Granulomatous arthritis represents the most frequent BS and EOS manifestation, occurring in almost all patients.20–22 Articular involvement shows mainly as symmetrical polyarthritis involving frequently proximal interphalangeal joints of hands and feet, ankles, knees and wrists.11 ,20–22 Arthritis of the metacarpophalangeal (MCP) joints and elbows is not infrequent, whereas the hips, spine and temporomandibular joints have been reported to be most rarely involved.11
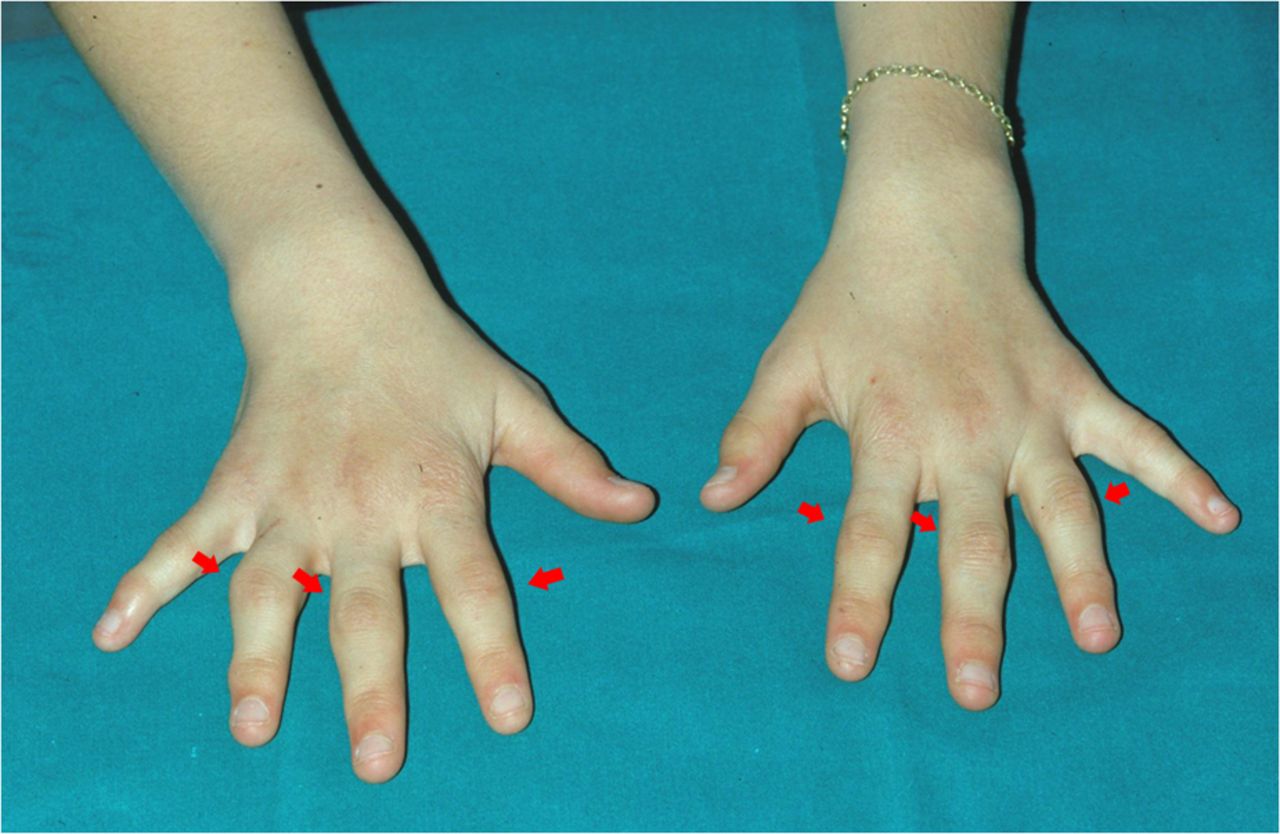
Joint involvement can be complicated by deformities; in particular, in acute phases, involvement of the hand small joints is often hypertrophic and associated with tenosynovial cysts (“boggy” synovitis and tenosynovitis) and flexion contractures9 ,11 (figure 2). Camptodactyly can also represent a common deformity in the early BS and EOS phases and be linked more to dysplastic processes and flexion contractures than to inflammatory processes.9 ,11 In a recent observational study by Rosé et al,11 X-rays of 45 hands have shown non-erosive aspects of arthritis, as well as in advanced diseases phases. In the same study, other significant radiological findings were represented by biconcave radial epiphysis, abnormal plump distal ulna and dysplasia of the scaphoid and lunate bone.11 Moreover, a high percentage of osteopenia and joint space narrowing was found at the level of the interphalangeal, MCP, radiocarpal and distal radioulnar joints.11
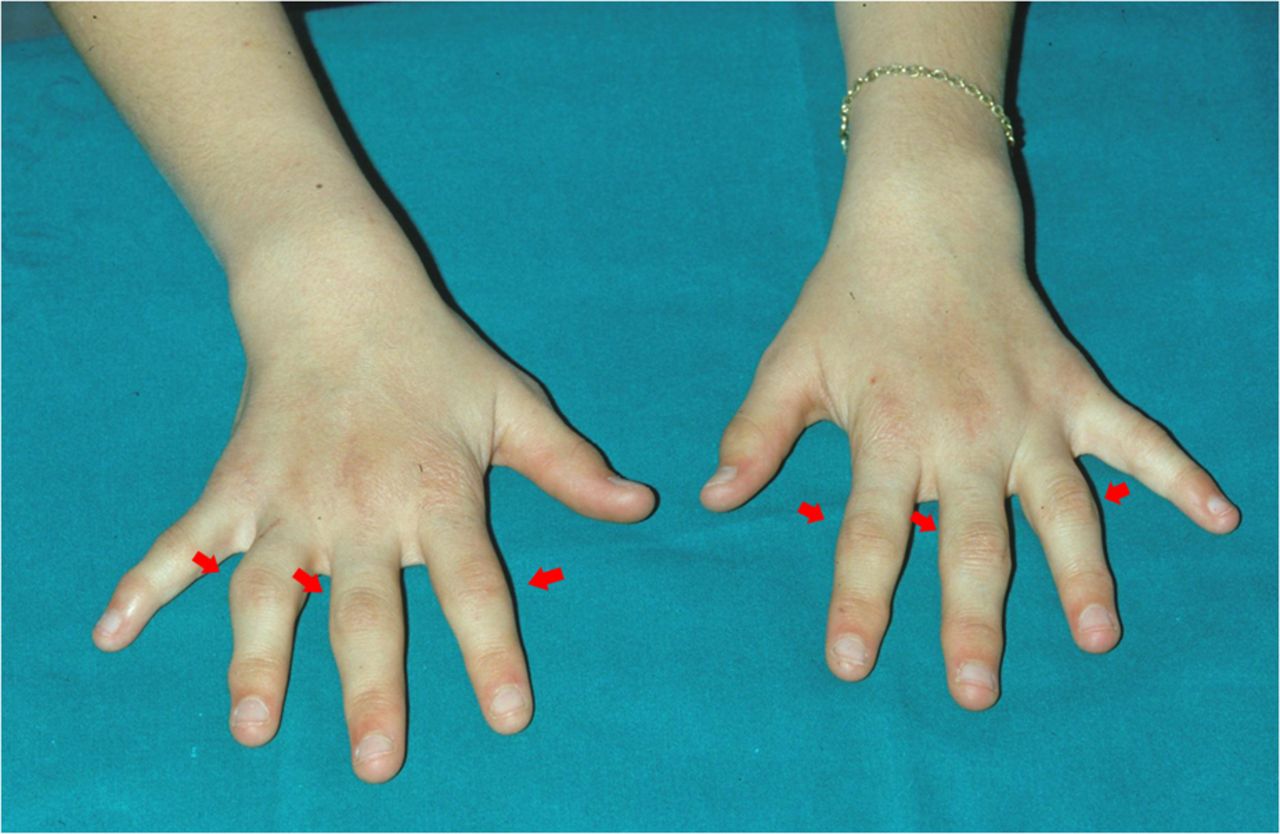
Camptodactyly in a young patient affected with BS.
Cutaneous findings
Dermatitis in the form of a dark red, slightly scaly, maculopapular, or eczematoid-like or lichenoid-like rash may occur frequently in the first year of life.9 ,11 Dermatitis is often symmetric, on the trunk and/or extremities and is usually intermittent with spontaneous resolution,. Other described cutaneous findings are represented by erythema nodosum, leucocytoclastic vasculitis, bilateral leg ulcers, ichthyosis vulgaris and pityriasis lichenoides.21 ,25–28
Ocular findings
Granulomatous uveitis is a frequent manifestation (70–80%), often bilateral and with a chronic recurrent course.11 ,29 Ocular granulomatosis is often referred to as blurred vision, painful and red eyes, floaters and photophobia.9 ,11 Ocular involvement can potentially evolve into moderate or severe visual loss, thus representing in many patients the most severe manifestation of these diseases.9 ,11
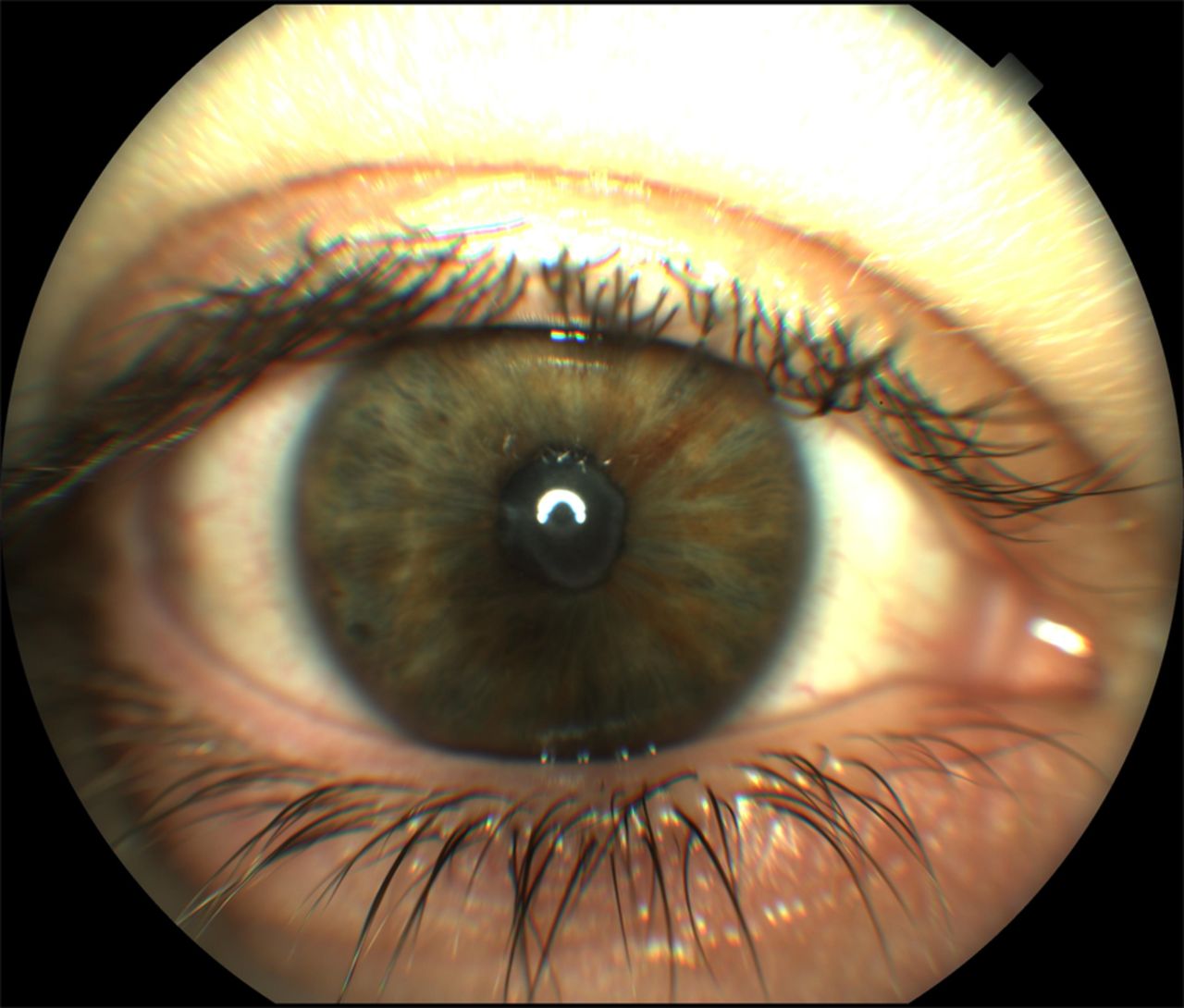
Uveitis can also affect all the ocular segments, with iridocyclitis, band keratopathy with inflammatory precipitates and vitriitis.9 ,11 Involvement of the posterior ocular segment with chorioretinitis, vasculitis, macular oedema with retinal detachment, and optic neuritis has been described in several cases.20–22 Cataracts, inflammatory glaucoma, peripheral synechiae, increased intraocular pressure and optic atrophy can represent uveitis complications.23 ,29 Figure 3 shows an acute flare of anterior uveitis in a patient with BS.
{kind=link}
{kind=link}
{kind=link}
Anterior uveitis in a patient with BS showing fibrin at the level of the anterior chamber.
Systemic and other body sites involvement
A heterogeneous spectrum of systemic manifestations has been reported in BS and EOS.20–22 Among these, recurrent fever has been reported as a frequent finding in patients carrying the p.R334Q/W mutation or other mutations, such as p.E383K.11 ,20–22 ,30
Neurological manifestations have been reported in familial and sporadic cases, especially in the form of cranial neuropathies and transient facial palsy.31 Recently, in a patient carrying a G481D mutation, in addition to transient facial palsy, ischaemic stroke has been described as an expanded manifestation.11 This patient also showed interstitial lung disease, nephrocalcinosis hypertension, lymphadenopathy and splenomegaly needing splenectomy.11 Acute transverse myelitis has been described in a patient with BS under therapy with the antitumour necrosis factor (TNF)-α, etanercept.32
In many patients, mainly carrying R334Q/W mutations, generalised lymphadenopathy, splenomegaly, hepatomegaly and/or hepatitis have been described.11 ,22 ,33–35
The presence of large vessels and Takayasu's arteritis have been reported in two patients with BS carrying p.R334W and p.G464W mutations, respectively.36
Takayasu arteritis has also been described in concomitance of cerebral haemorrhage, high fever with seizures, uveitis and acute pityriasis licheinodes in a Japanese patient with EOS carrying a D382E mutation.28 Arterial hypertension has been described in several reports.20 ,22
Renal involvement in the form of bilateral nephritis, nephrocalcinosis and chronic renal failure has also been observed.11 ,21 ,22 ,37 In several cases, the granulomatous pattern of nephritis has been confirmed by biopsy.22 ,38 The coexistence of renal cell carcinoma has been described in a Turkish patient with BS with the R334Q mutation, but it has not been clearly related to the disease.39
Heart involvement has been founded rarely associated with BS, as congestive heart failure, in a patient with G481D mutation; pericarditis has been described in two patients of the same family with BS carrying the R587C mutation.22 ,33 A case of left ventricular dysfunction and pulmonary haemorrhage due to bronchial granulomas has been reported.40
Sialoadenitis25 and granulomatous parotitis with a parotid gland swelling40 have also been found in different studies. Intestinal granulomatous inflammation has been described in a p.T605N carrier of a Norwegian family with BS.41
Differential diagnosis
Diagnosis of BS and EOS is mainly addressed by early onset, during the first years of life, of one or more recurrent manifestations of the granulomatous triad.3 The clinical picture needs to be corroborated by identification of possible CARD15/NOD2 gene mutations.42 Familial history is a key element in defining familial forms.42 Histological detection, obtained by biopsies of synovial and cutaneous lesions, can evidence the presence of non-caseating granulomas with epithelioid and multinucleated giant cells.12 Electron microscopy allows the identification of ‘comma-shaped bodies’ in epithelioid cells.12
Laboratory investigations exploring mainly autoimmune and infectious diseases are helpful to exclude mainly vasculitides, juvenile idiopathic arthritis, granulomatous infections and other granulomatous diseases.12 During inflammatory episodes, these investigations are required even in patients with a previously performed diagnosis of BS and EOS, in order to identify possible incoming overlapping comorbidities.12 Paediatric idiopathic sarcoidosis involves more frequently lungs and lymph nodes and is ruled out by CARD15/NOD2 gene analysis that showing absence of mutations exclude the genetic-driven nature of the inflammatory process.43 ,44
Therapy
High-dose corticosteroids have been shown to be efficacious when administered in concomitance with BS and EOS acute flares, as well as low-dose corticosteroids that are generally used during the quiescent stage.29 ,45 In refractory cases, the concomitant use of immunosuppressants (methotrexate, or azathioprine, or mycophenolate mofetil) has been shown to be effective.11 ,46 ,47 Further, the anti-TNF-α agent, adalimumab, in combination with corticosteroids and/or methotrexate, has recently been shown to induce remission in case of refractory arthrocutaneous, ocular and systemic involvement.48 Moreover, treatment with another anti-TNF-α drug, infliximab, has led to successful outcomes, also as monotherapy.11 ,49–51
If treatment with anakinra, an IL-1β receptor antagonist, has shown controversial results21 ,35 another IL-1β inhibitor, canakinumab, has been reported to be efficacious in improving severe uveitis.11 ,52
NOD2-associated autoinflammatory disease
In 2011, Yao5 et al described a new category of a complex autoinflammatory disease associated with CARD15/NOD2 polymorphisms and with phenotypic resemblance to BS.
This condition, recognised as NAID, was reported for the first time in seven adult Caucasian patients with symptoms of multiple system involvement, mean onset at above 40 years of age and no family history of other autoinflammatory diseases.5
The clinical picture was mainly characterised by febrile recurrent episodes, weight loss, fatigue, polyarthralgia/not erosive polyarthritis mainly with swollen and painful distal lower extremities, often associated with an increase in acute phase laboratory findings.5 ,53 ,54 In a 2013 study,53 data on clinical aspects were confirmed in an additional 15 patients.53 Skin manifestations were variable, providing mainly erythematous oedematous plaques, patches, macules, papules and linear rash.5 ,53 Granulomatous dermatitis with numerous histiocytes and several associated multinucleated cells was occasionally described.5 ,53
Gastrointestinal involvement, found in more than half of the patients, consisted of abdominal pain associated with diarrhoea and caecum-sigmoid colitis with non-necrotising granulomas.53 Ocular manifestations were represented by a Sicca-like syndrome53 and, in one case, ocular myositis.55 Recurrent chest pain, pleuritis and pericarditis were also described.5 ,53
All the described patients carried the IVS8+158 CARD15/NOD2 variant and, among these, several showed a concomitant p.R702W mutation.5 ,53
In addition, heterozygous p.T189M and p.R703C variants have also been reported to be associated with NAID.54 ,56 In particular, p.T189M has been found in an adult patient with recurrent fever, itchy dermatitis and polyarthralgia with painful swelling of the ankles, whereas p.R703C was found in a 63-year-old woman with dermatitis, arthritis, pneumonitis showing non-necrotising granulomas and diffuse interstitial lymphoplasmacytic infiltrates with multinucleated giant cells.54 ,56 Evidence of a triple dose of CARD15/NOD2 polymorphisms (IVS8+158, L1007fsinsC and R709Q) has been reported in an NAID adult patient with recurrent episodes of abdominal pain and high fever, multiple lymph nodules, dermatitis in association with eosinophilia and marked expansion of the submucosal ileum lymphoid tissue.57
Crohn's disease
CD is a chronic and recurrent inflammatory bowel disease, which can involve any part of the digestive tract, from the mouth to the anus, but mainly affecting the distal ileum and the colon.58 ,59
CD clinical course mainly depends by site and severity of inflammatory involvement, ranging from localised intestinal forms to diffuse and severe phenotypes.58 ,59 Its diagnosis is based on combined endoscopic, radiologicaland histopathological findings.58 ,59 The hallmark of active CD is represented mainly by an inflammatory cell infiltrate (lymphocytes, plasma cells) with focal crypt irregularity and independent granulomas.58 ,59
Abdominal pain, cramping, diarrhoea, blood and/or mucus in stool, nausea, vomit and weight loss represent the most frequent clinical findings and can be associated with systemic symptoms such as fever and fatigue.58 ,59 Concomitant autoimmune diseases and/or extraintestinal manifestations, as spondiloarthritis, uveitis, episcleritis and mucocutaneous features can also verify in course of CD.58 ,59 Therapeutic strategies are individualised and based on inflammatory location, severity, associated complications and systemic manifestations, providing mesalamine, sulfasalazine, disease-modifying antirheumatic agents and anti-TNF-α therapy.58 ,60 ,61
In 2006, McDermott and McGonagle have proposed CD as a polygenic disease, localised on the borderline between autoinflammatory and autoimmune disorders with a prevalent inflammatory component.62 Successively, the concept of CD as an autoinflammatory condition was reinforced by Masters et al,2 who classified this disease as an NF-κβ activation disorder, together with BS and NLR pyrin domain containing 2 (NLRP12)-associated autoinflammatory disease. To date, the exact aetiology of CD remains unknown, and the most accredited ethiopathogenetic hypothesis suggests a complex interaction between the predisposing genetic background and different components represented by the gut commensal microbiome and immune, environmental and risk factors.63 ,64 Among different genetic variants investigated in CD, three SNPs within LRR domains of the CARD15/NOD2 gene, p.R702T, p.G908R and p.L1007fsinsC, have been identified as susceptibility loci associated with the disease.4 ,65 Although the role of the NOD2 protein in CD ethiopatogenesis needs to be clarified, it is hypothesised that NOD2 susceptibility loci can contribute to reduced expression of α-defensin synthesised by intestinal Paneth cells and to loss of function, defective NF-κB activation and altered intestinal bacteria clearance with sequent changes in the gut microbiome.66–69
Other genetic associations have been identified in genes codifying interleukins and their receptors, such as IL17/23 axis molecules, and in genes codifying tyrosine protein kinase, signal transducer activator transcription phosphoproteins, involved in T-lymphocyte signal transduction.70 ,71 The gene codifying autophagy-related 16-like1 (ATG16L1) protein, which promotes autophagy, has been reported to be strongly associated with CD.72
Conclusions
The improvement of knowledge in molecular mechanisms and diagnosis of autoinflammatory diseases has represented an important breakthrough for improving their outcomes.73–76 With regard to BS and EOS, mainly thanks to International Registries and a 3-year observational study, there have been significant advancements in their understanding.11 ,20–22 Further, to date, the enhanced number of studies on the CARD15/NOD2 gene has allowed the identification of another NOD2-related disease, named NAID, resembling in part BS and EOS phenotypes. However, since this disease is still rather new and rarely described, data on clinical and molecular aspects are poor.5 ,53 ,54 Presence of non-caseating granulomatous pattern and inflammatory features and NOD2 susceptibility of CD has allowed then to portray this disorder as a granulomatous disease with an important autoinflammatory component. However, taking into account its heterogeneous genetic and clinical aspects, CD represents a disease located on the borderline between the autoinflammatory and autoimmune sides.2 ,62
The complex pathogenic mechanism behind granulomatous inflammation requests further studies in order to achieve complete models of disease pathogenesis and translating these into better patient outcomes.
References
Footnotes
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.