Article Text

Abstract
The aim of the review is to highlight the current knowledge about established and new biologicals and to summarise recent advances by focusing on comparative efficacy, safety and possible discontinuation of treatment in patients with rheumatoid arthritis (RA). Up to now, comparative analyses showed only minor differences with respect to efficacy and safety among the established biologicals. Studies confirmed the excellent drug retention rate as well as efficacy and safety of approved biologicals including their use in monotherapy. Tapering and in some instances discontinuation of biologicals is possible in disease remission. In case of relapse, patients usually show full response after reintroduction of the same compound. The development of biologicals continues fast with several new biologicals targeting different or established cytokines or cellular subsets of the immune system. With several new biologicals in the pipeline and different formulations for established compounds, treatment options for RA will become even more versatile and sophisticated. Although we get closer to the aim of decreasing the proportion of refractory patients, many questions have to be addressed in the near future regarding emerging biosimilars and biologicals with new modes of action.
- Rheumatoid Arthritis
- Treatment
- DMARDs (biologic)
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
Biological disease-modifying antirheumatic drugs (DMARDs) have translated the knowledge on molecular pathways into targeted therapies and are increasingly used in patients with rheumatoid arthritis (RA) with excellent efficacy and acceptable safety.
Head-to-head studies confirm comparable efficacy of different biological DMARDs in RA treatment, however, with respect to adalimumab monotherapy the results are favourable for tocilizumab.
Discontinuation studies show that patients with RA in sustained remission can successfully taper and sometimes stop tumour necrosis factor inhibitor without functional or radiological deterioration and, in case of relapse, restarting of biologicals is possible and regularly leads to rapid improvement.
Several new biologicals are in developmental status with the potential to further decrease the proportion of refractory patients.
Introduction
Disease-modifying antirheumatic drugs (DMARDs) are the mainstay of rheumatoid arthritis (RA) therapy. Institution of conventional synthetic DMARDs (csDMARDs) in therapy was followed by the development of tumour necrosis factor (TNF) inhibitors (TNFi), the first biological DMARDs (bDMARDs) introduced into rheumatology. Today, five different TNFi (infliximab (IFX), adalimumab (ADA), etanercept (ETN), certolizumab (CZP) and golimumab (GLM)) are in use. Although distinct by structure, route of application and pharmacokinetics, they show overall excellent effects with respect to clinical and radiological outcomes especially when used in comedication with methotrexate (MTX). TNFi are effective in all stages of disease including MTX-naïve patients with early RA and in patients with an inadequate response to MTX (MTX-IR) or any csDMARDs (csDMARD-IR).
Following TNFi, new bDMARDs with different modes of action were developed. Abatacept (ABT), targeting the co-stimulation between T and B cells, rituximab (RTX), targeting CD20+ B cells and finally tocilizumab (TCZ), an interleukin 6 receptor (IL-6R) antagonist, confirmed their efficacy in active RA including patients with an inadequate response to TNFi (TNFi-IR).
Strategy trials
Clinical studies with a predefined strategy can teach us a lot about the best treatment approach by using different available compounds. Furthermore, they are usually designed to answer relevant issues of daily practice.
Add-on strategies
In a large phase IIIb trial (REALISTIC), patients with RA with an inadequate response to at least one DMARD were randomised to receive CZP or placebo plus current therapy.1 The primary end point (week 12 American College of Rheumatology 20 (ACR20)) was met, and differences were already evident at week 2. Of note, the disease activity and physical function improved both in patients with or without previous TNFi use, regardless of their baseline MTX use.
A recently performed, open-label, prospective study (GO-MORE) evaluated the efficacy and safety of subcutaneous GLM as add-on therapy in csDMARD-IR patients with active RA.2 In this large study with 3366 patients, 82.1% achieved good-to-moderate European League Against Rheumatism (EULAR) responses and 23.9% attained remission at month 6.
In the ACT-RAY study, MTX-IR patients with active RA were randomised to add-on TCZ or to switch to TCZ plus placebo.3 After 1 year there was a trend favouring add-on strategy, however, both strategies demonstrated meaningful clinical and radiographic responses.
The aforementioned studies confirm efficacy of add-on strategies with biologicals in csDMARD-IR patients with RA, however, for TCZ, monotherapy also has convincing results.
Early aggressive treatment
The TEAR study has been designed to answer whether early aggressive treatment is comparable to a step-up approach in early RA.4 Patients were randomised to receive ETN+MTX or triple therapy (TT) consisting of MTX, sulfasalazine and hydroxychloroquine, or MTX-monotherapy. If low disease activity (LDA) was not reached, patients with MTX-monotherapy were allowed to advance to one of the combination therapies. Finally, the clinical outcomes were similar between all groups, but only the ETN+MTX group showed a significant radiological benefit. Results of a similar study with IFX (Swefot) were also consistent with these findings.5 These two studies support the current strategy of a step-up therapy beginning with MTX in patients with early RA.
TT versus TNFi+MTX in MTX-IR patients with RA
The RACAT study was designed to answer the critical question of whether TT is equivalent to ETN+MTX in patients with RA after MTX failure.6 In this 48-week, double-blinded, non-inferiority trial, patients were randomised to one of the arms, and if patients did not improve at 24 weeks they were switched to the other arm. After 24 weeks, both groups of patients improved significantly (p=0.001) with a switch rate of 27%. Of note, the degree of significant improvement and response rates were similar between both groups after switching. The primary outcome, changes in disease activity score 28 (DAS28), at 48 weeks, was similar in both groups (−2.1 with TT and −2.3 with ETN+MTX, p=0.26) suggesting non-inferiority for TT compared with ETN+MTX. However, the higher hurdles such as ACR50, ACR70, LDA and radiological progression were in favour of ETN. The study can be criticised for several methodical issues including change of outcome parameters and potentially reduced power due to non-achieved recruitment goal.
Role of MTX dose in combination with bDMARDs
In the CONCERTO trial, biological and MTX-naïve patients with early RA were randomised to open-label ADA plus weekly blinded 2.5, 5, 10 or 20 mg MTX.7 With increasing doses of MTX, significant increasing trends were observed in the proportion of patients achieving the outcomes. Of note, differences comparing 10 and 20 mg MTX were minimal. ADA serum concentrations increased with ascending dose up to 10 mg MTX. The authors related this significant trend with an effect of MTX on ADA pharmacokinetic profile.
Head-to-head comparisons of biologicals
First eagerly awaited studies for head-to-head (H2H) comparisons of biologicals were performed recently. In the RED SEA trial, 125 adults with active RA despite treatment with two csDMARDs including MTX were randomised to add-on therapy with ADA or ETN.8 There was no significant difference between treatment arms.
The 2-year results of the AMPLE study, a H2H comparison of ABT and ADA in MTX-IR patients with RA, confirmed similar efficacy based on clinical, functional and radiographic outcome.9 Safety outcomes were comparable between both groups with fewer local injection site reactions (ISRs) and slightly lower discontinuation rate under ABT.
Another H2H trial, ADACTA, compared monotherapy with TCZ versus ADA in patients with RA who were intolerant or inappropriate candidates for MTX.10 Of note, mean DAS28 improvement was significantly higher in the TCZ (−3.3) than in the ADA group (−1.8) (difference −1.5, 95% CI −1.8 to −1.1; p<0.0001) from baseline to week 24, while safety findings were comparable between the treatment arms.
In summary, these studies confirm comparable efficacy of different bDMARDs in RA treatment. However, with respect to monotherapy, the results are favourable for TCZ.
Alternative routes of administration
Since different routes of administration allow for more flexibility, trials for subcutaneous versus intravenous biologicals were performed in recent years. The ACQUIRE trial compared efficacy and safety of subcutaneous ABT versus intravenous ABT in MTX-IR patients with RA with a non-inferiority design.11 At month 6, similar proportions of patients in both arms achieved the primary outcome of ACR20 response (estimated difference: 0.3%, 95% CI −4.2 to 4.8). Both formulations demonstrated equal efficacy and safety including similar patient retention rates (94.2% for subcutaneous ABT vs 93.8% for intravenous ABT).
In the GO-FURTHER trial, MTX-IR patients with active RA were randomised to receive GLM intravenously or placebo with background MTX.12 Significantly more patients on GLM achieved the efficacy outcomes compared with placebo. Adverse events (AEs) were similar, but serious AEs (most commonly infections) were reported more frequently under GLM+MTX (4.1%) than placebo plus MTX (2%).
The SUMMACTA study randomly assigned patients with RA with an inadequate response to DMARDs (DMARD-IR) (including TNFi in up to 20% of patients) to receive TCZ subcutaneously or TCZ intravenously in combination with csDMARDs.13 At week 24, non-inferiority of subcutaneous versus intravenous administration was confirmed by similar ACR20 response rates. The safety profile of subcutaneous TCZ was consistent with the known safety profile of the drug with the exception of a higher incidence of ISRs.
Thus, in addition to ABT, intravenous and subcutaneous forms of TCZ and GLM are already available.
Discontinuation trials
Discontinuation of TNFi in MTX-naïve patients with RA
In recent years, several studies investigated the possibility of TNFi tapering or discontinuation. In a subanalysis of the BeST study, 64% of patients with initial IFX treatment and 25% of patients with delayed IFX exposure were able to discontinue IFX.14 Median time without IFX treatment was 17 months, and about 60% of patients paused IFX for at least 1 year. Restarting IFX resulted in DAS≤2.4 in all patients without any progression of radiological damage. Presence of shared epitope, smoking and a long treatment with IFX were independent predictors for IFX restart.
In another study published recently (PRIZE), DMARD-naïve patients with early active RA received 50 mg of ETN+MTX for 52 weeks and in case of qualifying responses were randomly assigned to receive 25 mg of ETN+MTX, MTX alone, or placebo for an additional 39 weeks.15 Patients with maintained responses after this period stopped all DMARDs and were followed to week 65. As a result, continuing combination therapy at a reduced dose led to better disease control than switching to MTX alone or placebo. However, radiological progression did not differ between groups.
In the OPTIMA study, 44% of MTX-naïve patients with early RA achieved LDA under ADA+MTX and were rerandomised to receive placebo plus MTX or to continue ADA+MTX.16 After 1 year, more patients with continuous ADA treatment maintained LDA or remission compared with MTX alone (LDA 91% vs 81%, p=0·0361; remission 86% vs 66%, p=0·0014).
Interesting data also came from the HIT HARD study, where patients with early RA were treated with ADA or placebo with MTX.17 After 24 weeks, both groups continued with MTX for a second period of 24 weeks. As a result, 47% of ADA+MTX patients achieved DAS28 remission, and 44% of these patients were still in remission at week 48. Despite similar results at 48 weeks, patients on ADA+MTX reached good clinical efficacy significantly earlier and had better radiographic outcomes.
Discontinuation of TNFi in csDMARD-IR patients with RA
The RRR study included patients with RA with persistent LDA for >24 weeks under IFX treatment.18 After IFX withdrawal, 56 of 102 patients maintained LDA over 1 year. In case of relapse, the majority again reached LDA by re-treatment with IFX. In fact, this study was the first to demonstrate the possibility of biological-free remission in patients with longer disease duration (mean 4.8 years).
The HONOR study recruited patients treated with ADA+MTX who agreed to discontinue ADA after sustained remission for ≥6 months (DAS28<2.6).19 It is noteworthy that in patients with deep remission (DAS28—erythrocyte sedimentation rate ≤1.98) identified by receiver operating characteristics analysis following logistic analysis in the study, after 1 year the proportion of patients with sustained remission or LDA was not significantly different between patients who continued or stopped ADA. In case of relapse, restarting ADA was effective and safe.
In the CERTAIN study, patients with RA with low-to-moderate disease activity were randomised to CZP or placebo plus current csDMARD.20 Patients who achieved remission stopped study treatment and followed up. In the follow-up, only 3 of the 17 prior CZP patients and 2 of the 6 prior placebo patients maintained remission until week 52. However, re-treatment with CZP was effective in relapsing patients.
Dose reduction of TNFi in MTX-IR patients with RA
In the PRESERVE trial, moderately MTX-IR patients with active RA pretreated with 50 mg of ETN+MTX were randomised to receive 50 mg ETN+MTX, 25 mg ETN+MTX, or placebo plus MTX.21 After 1 year of follow-up, in both treatment groups on ETN a higher percentage remained in LDA (82.6% and 79.1%, respectively) compared with MTX monotherapy (42.6%).
Discontinuation of TCZ in MTX-IR patients with RA
In the ACT-RAY study, 50.4% of patients discontinued TCZ following sustained clinical remission after 1 year.22 Subsequently, 84% of those patients experienced a flare up with recurrent response to the reintroduced drug.
Discontinuation of ABT in very early active RA
The AVERT study included patients with very early active RA who were randomised to subcutaneous ABT 125 mg plus MTX, ABT 125 mg monotherapy, or MTX.23 Patients with LDA at month 12 entered a second 12-month period of withdrawal of all RA therapies. A small but significant number of patients sustained drug-free remission in the ABT+MTX group compared with MTX alone at both 12 and 18 months (14.8% vs 7.8%, respectively; p=0.045).
Discontinuation of any DMARDs following sustained remission
The recently published prospective study, RETRO, analysed the effect of continuing, tapering or stopping DMARDs in patients with sustained remission.24 In total, 82.2% of patients received MTX, 40.6% received bDMARDs and 9.9% received other DMARDs. Overall, 66.3% of patients remained in remission for 12 months, whereas 33.7% relapsed. Of note, anticitrullinated protein antibody positivity and treatment reduction predicted relapse.
In summary, the available results show that patients with greater depth of remission are more likely to successfully taper and in some instances to stop bDMARDs. This is valid for patients with early as well as for patients with longstanding RA. Owing to limited results for non-TNFi, it is unclear whether differences with respect to drug-free remission exist for the available bDMARDs. However, in patients with relapse, restarting of biologicals regularly leads to rapid improvement.
New biologicals
Biosimilars
The patents for the earliest antirheumatic biologicals are expiring, which has encouraged the development of biosimilars. The IFX biosimilar CT-P13 has been approved by the European Medicines Agency. The phase III trial of CT-P13, PLANETRA, demonstrated equivalent efficacy to original IFX with a comparable pharmacokinetic and safety profile including immunogenicity.25 Clinical trials are also ongoing for ETN, ADA and RTX biosimilars. Intended copies of ETN and RTX are already in use in some countries.
Agents targeting IL-6
Apart from the approved bDMARDs, new biologicals targeting different or established cytokines or cellular subsets of the immune system are currently in clinical development for treatment of RA (figure 1).
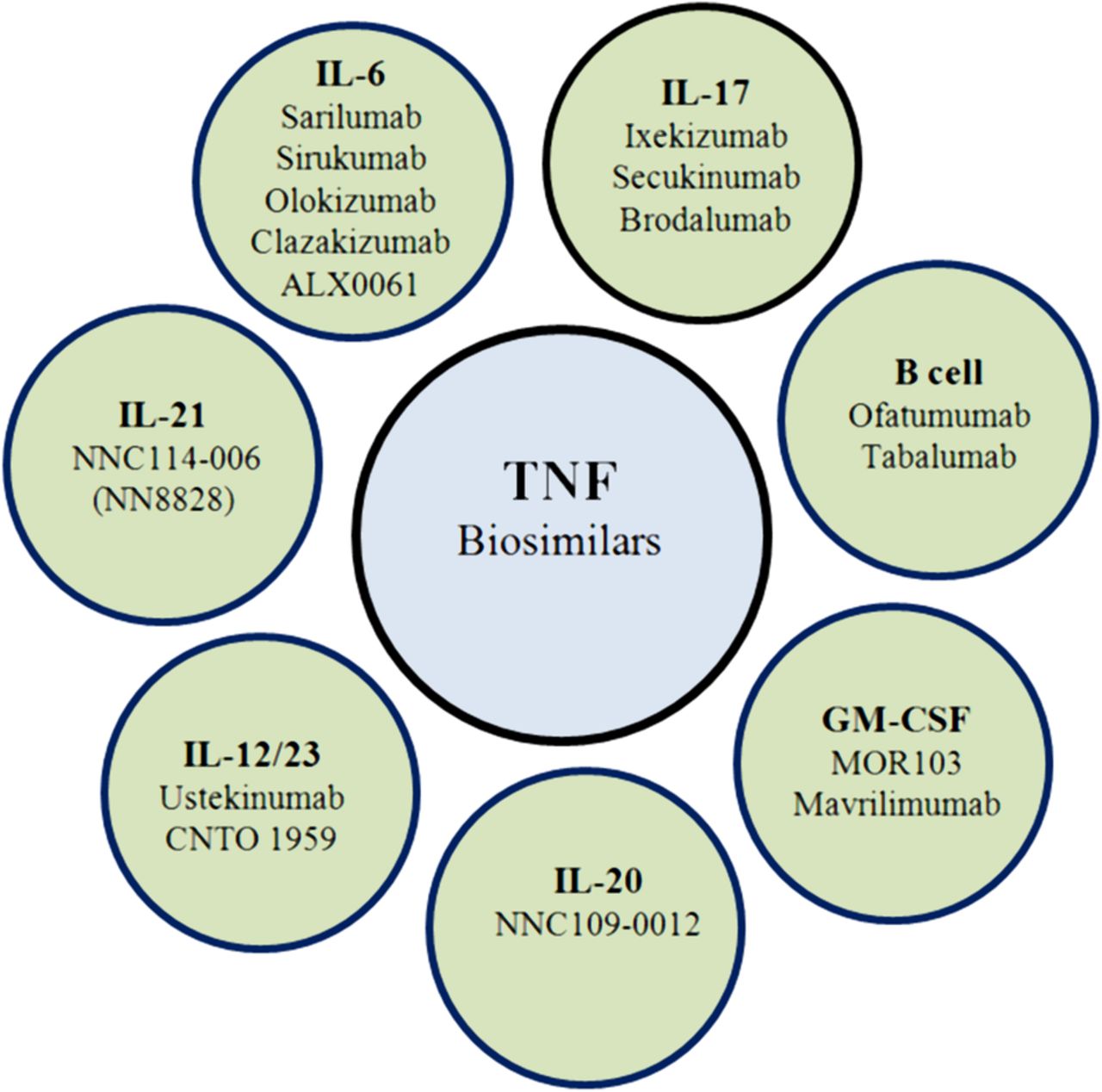
{kind=link}
New agents on developmental procedures targeting different cytokines or cells (GM-CSF, granulocyte–macrophage colony-stimulating factor; IL, interleukin; TNF, tumour necrosis factor).
Of these compounds, sarilumab, a fully human monoclonal antibody (mAb) against IL-6Rα was effective in MTX-IR patients with RA in phase II and III trials showing a safety profile similar to other IL-6 inhibitors.26 ,27 Published abstracts also confirmed a favourable efficacy–safety profile.28–30
Sirukumab, another new mAb targeting IL-6 also showed significant improvement in MTX-IR patients with active RA in a phase II trial.31 Safety results through 38 weeks were consistent with other IL-6 inhibitors and phase III trials are ongoing. Phase II trials with olokizumab and clazakizumab targeting IL-6 have also shown results consistent with other IL-6 inhibitors.32–34 However, with clazakizumab, no clear dose–response has been observed. Therefore, whether or not direct IL-6 inhibitors are really comparable to IL-6R antagonists has to be clarified by further studies. Another development in the field is the monovalent nanobody ALX-0061, also targeting the IL-6R. The data from a phase I/II study were promising with an 84% ACR20 response and 58% DAS28 remission rate.35
Agents targeting B cells
Beneficial results with RTX in RA have facilitated the development of several new drugs targeting B cells. Of these, two different humanised CD20 mAbs, ocrelizumab and ofatumumab, followed the same strategy of depleting B cells. Although ocrelizumab was shown to be effective, increased rate of serious infections led to termination of its development in RA.36–38 On the other hand, in a phase I/II study ofatumumab appeared to be effective and safe compared with placebo in DMARD-IR patients with RA.39 Subsequently, ofatumumab was also tested in biological-naïve MTX-IR patients with RA in a phase III study.40 ACR20 response rate was 50% versus 27% in the placebo group without any unexpected safety finding. A trial with subcutaneous ofatumumab in patients with RA on background MTX showed profound and prolonged B cell depletion without required glucocorticoid premedication.41
In addition to depleting strategies, neutralisation of B cell cytokines such as B cell-activating factor (BAFF) represents another alternative approach in RA. As an example, tabalumab is a fully human IgG4 mAb that neutralises membrane-bound and soluble BAFF. Although tabalumab showed some efficacy and acceptable safety in phase II trials,42–44 phase III trials could not confirm a clinical benefit.45–47
Agents targeting IL-17
In a phase I study, ixekizumab (LY2439821), a humanised IgG4 mAb against IL-17A, was effective in biological-naïve patients with RA, without significant safety signals.48 Recently published data of a phase II trial proved the same findings in patients with RA who were either biological-naïve or TNFi-IR.49 Furthermore, the published 1 year phase II data of another fully human IgG1k mAb against IL-17A, secukinumab, have also provided evidence that this treatment approach could be of benefit for csDMARD-IR and bDMARD-IR patients.50 The overall safety profile was acceptable with an increased rate of mostly mild infections of 31.9% and serious AEs in 8.9% of patients. Several phase III studies of secukinumab are ongoing, especially in TNFi-IR patients. In contrast, a phase Ib and a phase II study with brodalumab, a fully human IgG2 mAb against IL-17 receptor A (IL-17RA), showed good tolerance but no clinical relevant response.51 ,52
Agents targeting GM-CSF
Another cytokine with a close connection to the pathogenesis and clinical features of RA is granulocyte–macrophage colony-stimulating factor (GM-CSF). However, it was a long-time concern that targeted therapies against this cytokine could cause severe side effects such as neutropaenia or pulmonary alveolar proteinosis. Nevertheless, different compounds successfully entered clinical development for RA targeting the cytokine itself or its receptor. The phase I and phase IIa (EARTH) trials of mavrilimumab, a human mAb against GM-CSF receptor, showed profound and rapid onset of response, normalisation of acute phase reactants and an overall good safety profile.53 ,54 Moreover, the results of the phase IIb trial met the primary end points (DAS28 and ACR20) with a clear dose–response effect with excellent tolerability.55 Another compound is MOR103, a fully human mAb directed towards GM-CSF. This alternative approach was tested in a phase Ib/IIa study with promising results, including imaging data.56
Other agents
Ustekinumab, an anti-IL-12/23 p40 mAb, and CNTO 1959, a compound targeting IL-23 p19 were investigated in different regimes in patients with active RA on background MTX therapy in a phase II trial.57 The primary end point of ACR20 response was not reached; however, for improvement of DAS28 as a secondary end point, a significant difference was observed for patients receiving ustekinumab as well as for the higher dosage group of CNTO 1959.
Upregulation of IL-21 has been linked to increased disease activity and radiological damage in RA.58 Two phase I studies on safety, tolerability, pharmacokinetics and pharmacodynamics, and a phase II study regarding efficacy of a fully human anti-IL-21 mAb, NNC114-0006 (NN8828), have been completed, but results have not been presented so far.59–61
IL-20 and its receptors are present in RA synovial tissue and IL-20 is thought to play a role in the pathophysiology of RA.62 A novel human IgG4 mAb against IL-20, NNC0109-0012, was well tolerated in healthy respondents and patients with RA in phase I trials.63 ,64 Efficacy and safety results of phase IIa supported further trials.65 ,66 However, two phase II studies in MTX-IR and TNF-IR patients were terminated,67 ,68 and one phase II study investigating the mechanism of action through synovial biopsies was withdrawn prior to enrolment.69 Table 1 summarises selected new agents and their developmental phases.
Developmental status of new biologicals for RA treatment
A combined blockade of TNFα and IL-17 by bispecific anti-TNFα/IL-17 antibodies was tested in human mesenchymal cells and in an animal model of arthritis and was more effective than single blockade in inhibiting cytokine, chemokine and matrix enzyme responses, and in blocking tissue destruction.82 This observation may encourage new studies for combined blockade of appropriate cytokines.
Conclusion
Biologicals offer a new dimension in the treatment of RA, allowing us to translate the knowledge on the molecular pathways into targeted therapies. Today, bDMARDs are increasingly used in patients with an inadequate response or intolerance to csDMARDs. Excellent efficacy and acceptable safety of bDMARDs have been established in the previous years. They also possess the chance of tapering and discontinuation in patients with sustained remission. Beyond approved biologicals, several new biologicals are in developmental status with the potential to fill the current treatment gap and help us to crack the hard nut of patients with refractory RA.
References
Footnotes
Contributors All the authors contributed to the design of the work. ABA drafted the work, EF and G-RB revised it critically for important intellectual content. All the authors approved the submitted version of the manuscript.
Competing interests ABA has received honoraria for consulting or lecturing from BMS and Actelion. EF has received honoraria for consulting or lecturing from Roche/Chugai, BMS, Novartis, Pfizer, AbbVie and MSD. EF is currently receiving a grant from Organisation BMS and Novartis. G-RB has received honoraria for consulting or lecturing from Roche/Chugai, BMS, Novartis, Pfizer, AbbVie, UCB and MSD.
Provenance and peer review Commissioned; externally peer reviewed.
Data sharing statement No additional data are available.