Article Text

Abstract
Gout is one of the most severe and frequent rheumatic diseases. Clinical manifestations of gout arise from uric acid crystal deposition in the musculoskeletal tissue. At high concentrations of uric acid in the body (hyperuricaemia), needle-shaped monosodium urate (MSU) crystals are formed. The structures are ingested by neutrophils and monocytes and thereby trigger robust activation of the inflammasome, an intracellular protein complex mounting an inflammatory response. Inflammasome activation builds interleukin-1, which acts as a proinflammatory mediator and induces vasodilation, recruitment of additional leucocytes and the expression of proinflammatory cytokines and chemokines. This process is associated with the clinical manifestation of an acute gout attack. Such attacks, however, stop rather rapidly and the process of resolution of inflammation in gout is now better defined. Neutrophils having ingested MSU crystals undergo a specific form of cell death called NETosis, which is characterised by the formation of neutrophil extracellular traps (NETs). During this process, DNA is extruded, allowing the dense packaging of MSU crystals as well as the degradation of proinflammatory cytokines, thereby allowing the stopping of the inflammatory process. Reactive oxygen species are essential for forming NETs and for allowing the resolution of inflammation in gout. This process of NETosis is critical for understanding tophaceous gout, since tophi are composed of NETs and densely packed MSU crystals.
- Gout
- Arthritis
- Inflammation
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Precipitation of monosodium urate crystals in the tissue leads to an acute gout attack by inducing the inflammasome and releasing interleukin-1.
What does this study add?
Rapid resolution of inflammation in gout is based on the formation of neutrophil extracellular traps (NETs) resembling tophi.
How might this impact on clinical practice?
The data permit a better understanding of the mechanism of tophus formation and process of spontaneous resolution of inflammation in gout.
Introduction
Purine metabolism in apes and humans is fundamentally different from that in all other mammalian species. Whereas all mammals share the enzyme xanthine oxidase, which is requires to degrades xanthine, a product of the purine metabolism, to uric acid, the subsequent metabolic step mediated by the enzyme uricase, cleaving uric acid to allantoin, is lacking in apes and humans. The consequence of this loss of uricase is that the uric acid level is comparatively high in humans even at steady state conditions, as uric acid cannot be degraded and resembles the final product of purine catabolism. To allow the body to get rid of uric acid, which continuously accumulates in the body due to tissue turnover, excretion is preferentially managed by the kidney and, to a lesser extent, by the gut.
Hyperuricaemia and monosodium urate crystal formation
Conditions which increase uric acid levels in the body (hyperuricaemia), such as increased intake or production of purines, as well as impaired excretion of uric acid by the kidneys, bear the risk that uric acid cannot be kept in solution and starts to precipitate into crystals.1 Concentrations of uric acid over 6.4 mg per decilitre, which exceed its solubilisation limit, allow the precipitation of monosodium urate (MSU) crystals in the tissue. Increased dietary purine intake by, for example, excessive meat and beer consumption, or increased endogenous purine production associated with cell and tissue catabolism during forced diet or tumour cell lysis, are well-known conditions which increase the uric acid level in the body and facilitate the precipitation of MSU crystals. In addition, decline in kidney function due to ageing, disease or drugs, such as diuretics and aspirin, impairs the appropriate excretion of uric acid and therefore also increases the risk for crystal deposition. On the other hand, pharmacological interventions2 are able to lower serum uric acid levels in the body, thereby regaining solubility of uric acid and resolving even already existing MSU crystal deposits.3 Such measures include those which (1) effectively lower the production of uric acid such as the xanthine oxidase inhibitors allopurinol and febuxostat, or (2) those which directly cleave uric acid, such as the recombinant enzyme pegloticase, or (3) those fostering the excretion of uric acid through the kidneys, such as benzbromarone or lesinurad (figures 1⇓–3).

Mechanisms of acute gout attack. Needle-shaped MSU crystals (yellow) are ingested by phagocytes such as monocytes. The high sodium content of the crystals increases intracellular sodium concentrations (yellow dots). To maintain iso-osmolarity, water enters the cell through aquaporins and induces the swelling of the cells. Aside from the dilution of sodium in the cell, water also dilutes potassium, which falls below a critical level for the induction of the inflammasome. Activation of the inflammasome produces large amounts of interleukin 1 β (aggNETs, aggregated neutrophil extracellular traps; MSU, monosodium urate; NETosis, aggregated neutrophil extracellular trap formation).

Mechanisms of chronic tophaceous gout. Neutrophil granulocytes ingest needle-shaped MSU crystals (yellow). During low concentrations of neutrophils, this process releases cytokines (red dots) and recruits further cells. On ingestion of MSU crystals, neutrophils undergo a specific form of cell death known as NETosis. NET resembles extracellular DNA. The extrusion of DNA during NETosis densely packs MSU crystals and cytokines. The high number of neutrophils at sites of MSU deposits allows the formation of aggregated NETs, which are highly effective in degrading cytokines (IL-1, interleukin 1; MSU, monosodium urate; NET, neutrophil extracellular trap).
{kind=link}
{kind=link}
{kind=link}
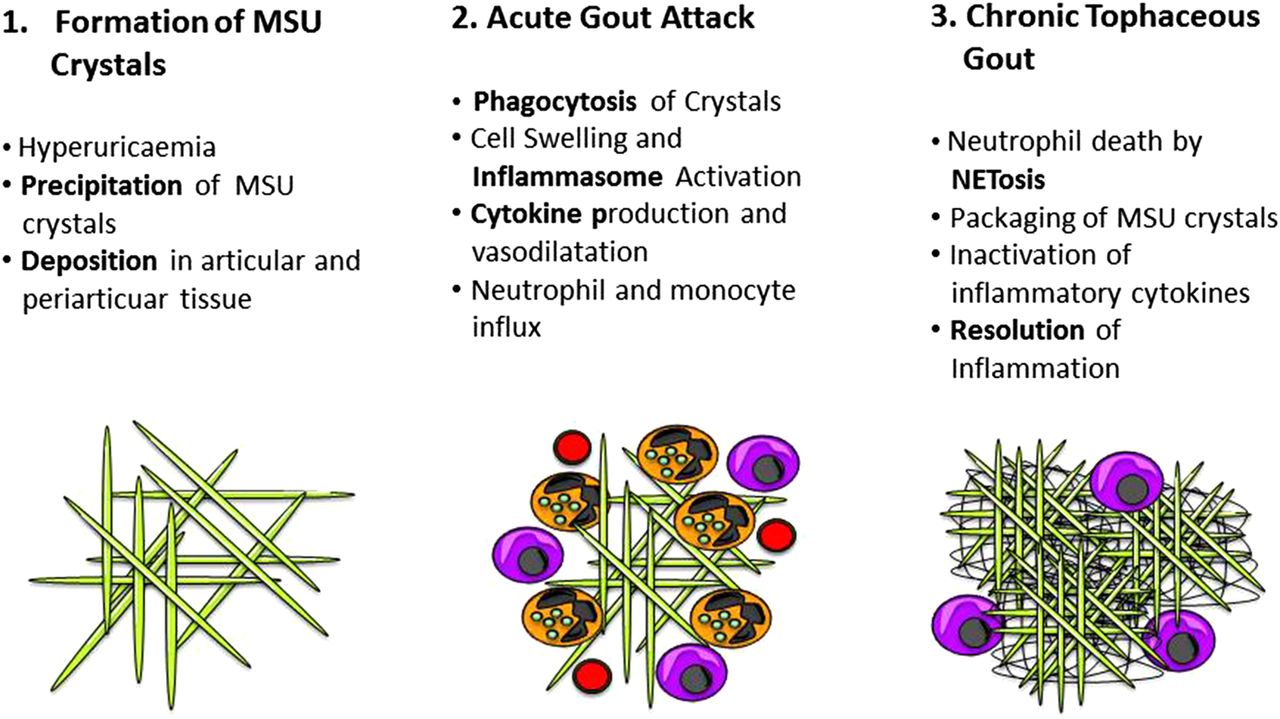
Immunopathogenesis of gout. Phase 1 shows the precipitation and deposition of needle-shaped monosodium urate (MSU) crystals. Phase 2 shows the acute gout attack with neutrophils and monocytes ingesting MSU crystals and releasing cytokines. Phase 3 shows the formation of a tophus during tophaceous gout showing neutrophil extracellular trap formation (NETosis).
Pathogenesis of acute gout attack
MSU crystal formation and deposition is the key pathogenic process of gout. MSU crystals are needle-shaped structures that are rapidly recognised by human phagocytes and ingested. Owing to the high concentrations of sodium in the crystals, ingestion of such structures by phagocytes abruptly increases the cellular sodium content.4 The consequence of this intracellular hyperosmolarity is the compensatory influx of water into the cytoplasm and cell swelling. Although this process restores normal osmolarity in the cell, it dramatically decreases the potassium content, which represents the key signal for the induction of the inflammasome and results in excessive production of interleukin 1 by those cells, which have ingested MSU crystals. IL-1 then promotes a massive inflammatory response with vasodilation and the rapid recruitment of immune cells, preferentially neutrophils to the site of crystal deposits.5 ,6 Other pathways such as toll-like receptor (TLR) activation by MSU crystals as well as free fatty acids add to the activation of the inflammasome and IL-1 production.7 This rapid and profound inflammatory response of the body to MSU crystal deposition manifests clinically as an acute gout attack. Importantly, the detection of phagocytes having ingested MSU crystals is still the gold standard clinical procedure to identify gout and also reflects the causal relationship between MSU crystal formation and inflammation during acute gout.
Pathogenesis of chronic tophaceous gout
One of the most intriguing observations in gout is that inflammation resolves quite rapidly despite the sustained presence of the triggering factor, namely MSU crystals. In contrast to other forms of inflammatory disease affecting the musculoskeletal system, such as rheumatoid arthritis or spondyloarthritis, the symptoms of an acute gout attack cease after a few days, suggesting that the body mounts effective mechanisms to stop inflammation. It needs to be mentioned that inflammation does not resolve because it effectively clears MSU crystal deposits and hence removes the triggering factor. In fact, the body manages to turn down inflammation despite the fact that MSU deposits are still present, which is a remarkable process. Imaging studies by dual-energy CT have impressively underlined this finding and have shown that profound MSU crystals deposits can be observed in articular and periarticular structures despite the absence of clinical signs of inflammation.8 ,9 Clinically, this stage of the disease is known as chronic tophaceous gout, which can be clinically silent for rather longer periods before a flare reoccurs.10
Neutrophil extracellular traps and the nature of tophi
Until recently, the reason for the spontaneous rapid resolution of inflammation in gout was rather enigmatic. While anti-inflammatory mediators, such as transforming growth factor β, melanocortin and IL-10, as well as a switch from proinflammatory to anti-inflammatory macrophages have been discussed as potential factors responsible for the resolution of inflammation in gout,11–14 these concepts rather refer to common anti-inflammatory mechanisms rather than characterising specific mechanisms involved in the resolution of inflammation in gout. Recent investigations of human tophi, which resemble densely packed MSU crystal deposits, however, revealed the abundant presence of extracellular DNA in these structures.15 Moreover, the extracellular DNA is in direct contact with the MSU crystals providing kind of a “trap” for these pathogenic structures and allowing their dense packaging.16 Mechanistic studies uncovered the process how these large amounts of extracellular DNA, found inside the tophi, form. Hence, ingestion of MSU crystals by neutrophils, which are by far the most abundant leucocyte subset involved in gout, leads to NETosis of these cells. NETosis, standing for neutrophil extracellular trap formation, describes a specific process of cell death, which allows the rapid extrusion of DNA. The extruded DNA forms a trap allowing to neutralise bacteria as well as other danger signals such as MSU crystals.15 ,17
With increasing amounts of neutrophils recruited to the sites of MSU deposits, NETs increase and densely cluster the MSU crystals when forming aggregated NETs (aggNETs). These large structures can only form when neutrophil densities are high, which happens at the later stages of the gout attack, when sufficient amounts of neutrophils have already been recruited. Hence, aggNETs formation represents the tipping point when an active inflammation collapses and the resolution process starts. Taken together, gout tophi represent aggNETs containing large amounts of extracellular DNA with interspersed MSU crystals. These structures are part of the body's strategy to contain the detrimental effects of MSU crystal deposition by effectively packaging them in DNA.
Resolution of inflammation in gout
However, how do aggNETs effectively turn down inflammation? Functional studies with NETs have shown that these structures are highly potent to trap and cleave inflammatory cytokines within minutes. Hence, even the addition of small amounts of aggNETs to cell cultures effectively absorbs most of the proinflammatory cytokines and chemokines required to mount inflammation within a short time.15 Even more importantly, the proinflammatory mediators are trapped in these structures as well as are degraded and inactivated. Degradation of cytokines, such as tumour necrosis factor-α, IL-1 and IL-6, and chemokines, such as monocyte chemoattractant protein (MCP)-1 within aggNETs, thus removes the fuel of inflammation and the process of inflammation then rapidly collapses. Thereby, this process is guided by the large amounts of neutrophil proteases, which are exposed during the process of NETosis and which become highly enriched in the aggNETs.
The role of reactive oxygen species
It is interesting that NETosis and the resolution of inflammation in gout is an active process driven by the neutrophil granulocyte. This process essentially requires the formation of reactive oxygen species (ROS).15 ROS represent molecules, such as superoxide anions and peroxides, which contain oxygen and are highly chemically reactive. ROS are induced during environmental stress, a process which is mediated by (nicotinamide adenine dinucleotide phosphate) NADPH oxidases, also known as NOX enzymes.18 Neutrophil function, such as bacterial killing, as well as the process of NETosis, essentially depends on ROS.19 If ROS production is impaired, for example, by the induced mutation of NOX enzymes in mice or by mutation of the enzyme in humans, found in chronic granulomatous disease, NET formation is severely impaired.15 Supporting the key role of NETosis in inflammation, dysfunctional NETosis associated with NOX mutations does not allow proper resolution of inflammation. For instance, if mice with dysfunctional NOX and impaired NETosis are challenged with MSU crystals, they develop chronic arthritis lasting for several weeks instead of a spurious self-limited disease lasting only a few days. These observations underline the importance of aggNET/tophus formation in response to MSU deposits and explain why and how MSU crystal deposits can be tolerated without massive inflammatory response for a rather long time in the body.
Conclusion
Taken together, two essential molecular phases can be distinguished in gout: the first one is the precipitation of MSU crystals and their ingestion by mononuclear phagocytes leading to inflammasome activation. This molecular process, which depends on the production of IL-1 and is characterised by the massive recruitment of neutrophils, explains the clinical symptoms of an acute gout attack. The second phase is the formation of tophi resembling aggNETs, which depends on high densities of neutrophils, and their death by NETosis, which allows densely packing MSU crystals, neutralising and degrading the involved proinflammatory cytokines and resolving inflammation. This molecular process described the clinical picture of chronic tophaceous gout.
References
Footnotes
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.
Data sharing statement No additional data are available.