Article Text

Abstract
Osteoarthritis (OA), a whole-joint disease driven by abnormal biomechanics and attendant cell-derived and tissue-derived factors, is a rheumatic disease with the highest prevalence, representing a severe health burden with a tremendous economic impact. Members of the nuclear factor κB (NF-κB) family orchestrate mechanical, inflammatory and oxidative stress-activated processes, thus representing a potential therapeutic target in OA disease. The two pivotal kinases, IκB kinase (IKK) α and IKKβ, activate NF-κB dimers that might translocate to the nucleus and regulate the expression of specific target genes involved in extracellular matrix remodelling and terminal differentiation of chondrocytes. IKKα, required for the activation of the so-called non-canonical pathway, has a number of NF-κB-independent and kinase-independent functions in vivo and in vitro, including controlling chondrocyte hypertrophic differentiation and collagenase activity. In this short review, we will discuss the role of NF-κB signalling in OA pathology, with emphasis on the functional effects of IKKα that are independent of its kinase activity and NF-κB activation.
- Osteoarthritis
- Chondrocytes
- Inflammation
- Cytokines
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Osteoarthritis (OA) is the most common joint disease and the major cause of disability in the adult population; with annual costs of knee OA being immense, this continues to be a severe health burden with respect to morbidity and expense. Age is the primary OA risk factor, and ageing-related changes also contribute to pathophysiological changes triggering OA disease. In addition, individuals with other specific OA risk factors, including obesity, altered joint mechanical loading, joint injury and inflammation, as well as genetic components,1 may undergo an accelerated rate of changes that are similar to those associated with ageing.2 Although cartilage destruction is the hallmark of OA, and collagen erosion is the pivotal event that determines the irreversible progression of OA disease, it is now well established that OA is not only a disorder of cartilage homeostasis but is a whole-joint disorder involving all joint tissues, including the subchondral bone, menisci and synovial membrane.3–5 In spite of recent advances, the mechanisms leading to cartilage destruction in patients with OA are still not clearly identified and no successful therapeutic intervention exists. This brief review elaborates on how nuclear factor (NF-κB) signalling contributes to OA pathophysiology, not only by modulating stress-related processes, but also by the hypertrophic-like conversion of OA chondrocytes.
Cartilage degradation in OA disease: phenotypic shift of articular chondrocytes
The principal function of articular cartilage is to adjust to biomechanical forces during joint movement; a function mediated by the extracellular matrix (ECM). Cartilage homeostasis is defined by normal cartilage ECM compensating for mechanical stress without structural or cellular damage. The ECM is produced and maintained by articular chondrocytes, the unique cell type residing in articular cartilage, which are essential for maintaining structural and functional integrity of the cartilage.6 Mechanical, oxidative and inflammatory stresses activate signal transduction pathways in cartilage, which disseminate a phenotypic shift characterised by the release of the chondrocyte from growth arrest, imbalanced homeostasis, hypertrophic-like conversion and aberrant expression of proinflammatory and catabolic genes.6 As a result, OA chondrocytes are unable to maintain tissue homeostasis and fail to replace ECM, especially collagen, once it is degraded by metalloproteinases (MMPs), in particular by MMP-13, the major MMP responsible for remodelling type II collagen in cartilage tissue.7–9 Characteristic features of OA chondrocytes in ageing and inflammatory models include prominent epigenomic alterations10 ,11 leading to deregulated gene expression, exacerbated and sustained NF-κB activation, and MMP production, all of which can be modelled in vitro by interleukin 1 (IL-1β) stimulation.12
The NF-κB pathway is most prominent among the different gene signatures in human disease and different experimental OA models, suggesting a key regulatory role for stress and inflammatory signalling via the canonical NF-κB pathway in OA pathology.6 ,12–14 Abnormal NF-κB activation provokes the loss of the growth-arrested state of articular chondrocytes, accompanied by the production of procatabolic mediators, including aggrecanases and MMPs that induce cartilage degradation, and the proinflammatory cytokines (IL-1β and tumor necrosis factor α,TNF-α) that induce them. Further, continued NF-κB activation results in the overexpression and activation of other regulatory transcription factors, including E74-like factor 3 (ELF3) and endothelial PAS domain protein 1 (hypoxia inducible factor 2 alpha) (HIF-2α), which, in turn, further perpetuate OA disease via modulation of inflammatory and catabolic mediators,15–17 and by linking hypertrophic-like conversion with inflammation.18 ,19 Together, these multilayered signalling networks collaborate to activate MMP13 expression and activity, and facilitate the progression of normal articular chondrocytes to a hypertrophic-like OA phenotype in vivo, thereby also contributing to OA onset and/or progression.6
Synovitis is a feature of the OA joint, in which the activated canonical NF-κB subunits upregulate the expression of chemokines (IL-8, chemokine (C-C motif) ligand 5 (CCL5)), cytokines (receptor activator of nuclear factor kappa-B ligand (RANKL), IL-1β, IL-6, TNF-α), cyclooxygenase 2 (COX2) and angiogenic factors (vascular endothelial growth factor, fibroblast growth factor 2).4 ,5 The homeostasis of the subchondral bone involves a balance between bone resorption, mediated by the receptor activator of nuclear factor kappa B (RANK)/RANKL signalling pathway that activates NF-κB transcription factors (inducing the expression of IL-1β, IL-6, prostaglandin E2 (PGE2)) and bone formation, controlled by osteoprotegerin (OPG). During OA, homeostasis is destroyed and the OPG/RANK/RANKL signalling pathway is deregulated.14
Contribution of the NF-κB pathway to cartilage degradation and OA pathology
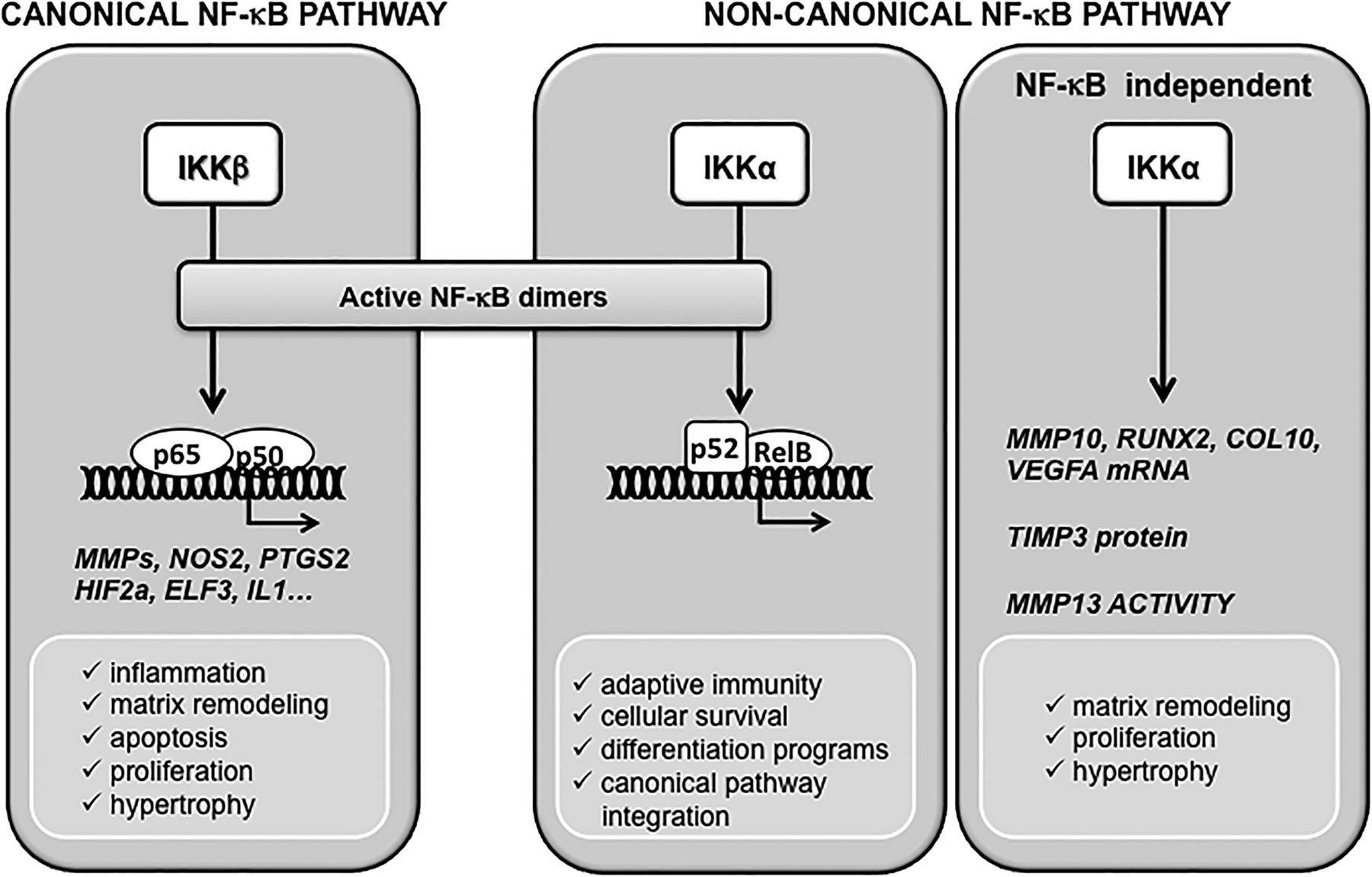
NF-κB-mediated transcriptional control arises from the assembly of homodimers and heterodimers of five different NF-κB proteins.20 NF-κB dimers are sequestered in the cytoplasm with their transcriptional activities blocked by inhibitory IκB proteins. The two pivotal kinases IKKα and IKKβ initiate the release of active NF-κBs from IκBs, so that specific NF-κB dimers might translocate to the nucleus and regulate the expression of specific target genes. In response to a host of proinflammatory and stress-like stimuli, IKKβ is the dominant acting IκBα kinase in vivo, controlling the so-called canonical pathway.20 However, IKKα is specifically required for the activation of the non-canonical, or alternate, NF-κB pathway, and has a number of in vivo functions as a serine–threonine kinase acting independently of NF-κB signalling (figure 1).12 ,21
In chondrocytes, IKKα and IKKβ have different functional roles in ECM remodelling and endochondral ossification, which are also developmental events recapitulated to a certain extent by hypertrophic-like chondrocytes in OA disease.22–24 Furthermore, in activated OA chondrocytes, the canonical NF-κB pathway is an orchestrator of gene expression programmes leading to the production of catabolic enzymes, cytokines and inflammatory mediators,12 ,25 and it is believed to play pivotal roles in OA disease by coordinating a complex, multilayered signalling network. Table 1 summarises some of the events controlled by IKKα and IKKβ in OA disease.

{kind=link}
Contribution of IKKα and IKKβ to chondrocyte pathophysiology. IKKβ and IKKα, the activating kinases of the canonical and non-canonical NF-κB pathways, respectively, contribute to maintain normal chondrocyte homeostasis and are major players in cartilage pathology. The IKKβ-controled canonical NF-κB signaling orchestrates most stress/inflammatory responses and modulates, among others, hypertrophy or matrix remodeling, either directly or via downstream mediators including HIF2α or ELF3. The non-canonical NFκB signaling, controlled by IKKα, mediates adaptive immunity and participates in cell survival and differentiation processess in different cell types. In chondrocytes, IKKα mediates chondrocyte hypertrophic differentiation and MMP13-driven collagenase activity in a kinase-independent manner in vitro. These processes have implications in osteoarthritis disease, where abnormal chondrocyte phenotype, enhanced collagenase activity, and the imbalance between anabolism and catabolism, lead to irreversible extracellular matrix degradation and cartilage destruction.
Summary of the contribution of IKKα and IKKβ to chondrocyte catabolism and cartilage degradative processes
Control of inflammatory/stress responses by the canonical NF-kB pathway
The IKKβ-driven, canonical NF-κB pathway signalling also has effects on downstream regulators of terminal chondrocyte differentiation (including HIF-2α, β-catenin and Runx2), thus linking inflammatory and oxidative stress responses, with phenotypic and functional changes in OA chondrocytes. These events further deregulate chondrocyte homeostasis and normal function (reviewed6 ,12). In OA chondrocytes, canonical NF-κB signalling acts as a sensor of exogenous and endogenous mechanical stress and inflammatory insults, and thereby coordinates the expression and activation of an abnormal cartilage catabolic pathway leading to the onset of OA disease (see reviews6 ,12), which also includes modulation of mitogen-activated protein kinase signalling via transcriptional control of GADD45β.44–46 In addition, NF-κB signalling plays a central role in disease progression and perpetuation, mediating a cascade of inflammatory responses triggered by advance glycation end products, Toll-like receptor ligands, or released ECM products, including fibronectin fragments, which lead to the continued expression of MMPs, aggrecanases, inflammatory cytokines and chemokines, and to an abnormal differentiation status.26–30 ,33–41 ,47
Among the downstream effectors of IKKβ-driven NF-κB signalling that play a central role in OA disease by coordinating a complex, multilayered signalling network, is HIF-2α (for review, see ref 12). HIF-2α has been shown to have central roles in OA disease by interconnecting inflammatory ECM degradative processes with chondrocyte hypertrophic conversion due to its control of genes involved in endochondral ossification18 and inflammatory factors,19 and its ability to trigger MMP13 via concerted actions between CCAAT-enhancer-binding protein beta (C/EBPβ) and RUNX2.32 ELF3 is another direct NF-κB target with central roles in cartilage catabolism and OA disease. ELF3 is an epithelium-specific member of the E26 transformation-specific sequence family of transcription factors49 with functional roles in epithelial cell differentiation, apoptosis and gut development.49–51 More recently, ELF3 has been shown to participate in a feedback loop with NF-κB signalling, by constitutively activating and stabilising canonical NF-κB in prostate cancer.52 In response to stress/inflammatory stimuli and depending, at least in part, on canonical NF-κB (p65/p50) signalling, ELF3 is induced in different tissues and cell types, where it mediates inflammation by controlling the expression of nitric oxide synthase 2 (NOS2) or COX2, among other targets.31 ,53 In chondrocytes, ELF3 expression is increased in OA cartilage and induced in vitro by IL-1β, thereby contributing to the IL-1β-mediated repression of the collagen, type II, alpha 1 (COL2A1) promoter16 and the activation of MMP13 transcription.15
NF-κB-independent actions of IKKα in articular chondrocytes
During OA, chondrocytes undergo phenotypic modulation, and display increased expression and activities of matrix-degrading enzymes, and abnormal production of matrix structural proteins, including type X collagen. Importantly, factors that drive chondrocyte hypertrophy, including Notch signalling, indian hedgehog (IHH), RUNX2 and HIF-2α, have all been implicated in OA disease.12 ,18 ,19 ,32 ,54–58 The knockdown (KD) of either IKKα or IKKβ in three-dimensional cultures of human articular OA chondrocytes revealed essential and differential roles for IKKα and IKKβ on ECM remodelling and terminal differentiation.42 While the contribution of IKKβ predominantly involved downregulation of SRY (sex determining region Y)-box 9 (SOX9) expression42 and activity,43 surprisingly, IKKα KD led to a more pronounced alteration of the hypertrophic-like conversion of articular chondrocytes. The KD of IKKα (IKKα KD) dramatically stabilised the ECM, enhanced cell viability and strongly suppressed chondrocyte differentiation towards a hypertrophic-like state. Importantly, while IKKα loss did not modify MMP-13 protein levels, it did inhibit collagenase activity, as revealed by the markedly suppressed accumulation of type II collagen fragments containing the 3/4C neo-epitope (COL2–3/4C).42 Importantly, the functional effects of IKKα on ECM remodelling, and subsequent aspects of chondrocyte differentiation towards a hypertrophic-like state, are evolutionarily conserved between human OA chondrocytes and immature murine articular chondrocytes, indicating that these properties of IKKα are intrinsic to chondrocytes and independent of other acquired phenotypes specifically linked to OA-affected cartilage.48 IKKα does not regulate MMP-13 gene expression, but positively modulates ECM remodelling by directly upregulating the messenger RNA (mRNA) encoding the procollagenase activator MMP-10 and by post-transcriptionally suppressing TIMP (tissue inhibitor of MMP) 3 protein levels. Together, these processes maintain maximal MMP-13 activity, which is required for ECM remodelling leading to chondrocyte differentiation. Thus, IKKα directly modulates total collagenase and MMP-13 activities both in human OA and in differentiating primary murine chondrocytes.
Previous evidence revealed that IKKα is essential for keratinocyte differentiation in murine embryonic development,59 ,60 but independent of NF-κB activation and its kinase activity.61 The abnormal skeletal development of IKKα knockout (KO) mice was due to failed epidermal differentiation, which disrupted normal epidermal–mesodermal interactions.62 Subsequent molecular analysis uncovered the role of IKKα as a critical regulator of Smad4-independent TGFβ-Smad2/3 signalling that directly induces c-Myc antagonists, which are required for terminal epidermal cell differentiation.63 Interestingly, we also found that kinase-independent functions of IKKα are required for primary chondrocyte differentiation towards hypertrophy.48 Thus, rescuing IKKα expression in both IKKα KD human OA chondrocytes and IKKα KO murine chondrocytes using viral transduction of murine wild type IKKα or a recombinant IKKα kinase-dead mutant, IKKα(K44M), fully restored collagenase activity and the ability of articular chondrocytes to differentiate towards hypertrophy.48 Detailed analyses revealed that the enforced expression of a kinase-dead IKKα mutant (even at physiological protein levels) completely rescued the expression of markers of hypertrophy, including Runx2 and Col10a1, to wild-type levels. Importantly, the kinase-dead mutant also reversed the accumulation of TIMP-3 in IKKα KD human OA chondrocytes and the suppression of MMP10 mRNA and protein levels in IKKα KO murine chondrocytes, both of which were associated with the recovery of type II collagen remodelling in these primary articular chondrocytes. Taken together, these observations represent strong evidence that the functional effects of IKKα in chondrocytes are independent of its serine–threonine kinase activity and thus also independent of the IKKα-dependent non-canonical NF-κB signalling pathway.48 Interestingly, TGFβ-Smad2/3 signalling has also been reported to positively regulate MMP-10 transcription in breast cancer epithelial cells, but a role for IKKα in this context was not determined.64 Future work will, in part, determine if IKKα, in a manner independent of its kinase activity, operates via TGFβ-dependent Smad2/3 signalling to activate MMP-10 transcription and simultaneously suppresses TIMP-3 levels in differentiating chondrocytes.
Conclusion
A better understanding of the mechanisms involved in the initiation and progression of OA disease stands essential for the development of effective, specific and successful treatment options for the disease, as well for the identification of predictive biomarkers that allow us to identify early stages of disease or individuals at high risk of developing OA. The IKKβ-driven canonical NF-κB signalling pathway coordinates mechanical, inflammatory and oxidative stress-activated events leading normal chondrocytes to become activated and hypertrophic-like. In vitro experiments with primary chondrocytes indicate that IKKα also functions as a positive mediator of this process independent of its kinase activity. Thus, controlling the mechanisms of action of IKKα or IKKβ holds potential for the development of therapeutic strategies to attenuate the onset and/or progression of OA disease pathology.
Novel strategies are being developed by selective inhibition of key molecules in the NF-κB signalling, in particular the canonical activation of RelA/p65 (by using small interfering RNA targeted to the RelA/p65 mRNA) or p50 (via annexin A4 modulatory effects).14 Indeed, non-specific therapeutic approaches that affect the NF-κB pathway are available, such as non-steroidal anti-inflammatory drugs, glucocorticoids, glucosamine, thalidomide and nutriceuticals, such as curcumin and sirtuin,65 as well as, potentially, anticytokine therapies such as dual acting anti-IL-1β/α, IL-1 receptor antagonists and anti-TNF-α. Novel strategies are being developed by selective inhibition of key molecules in the canonical NF-κB pathway. One in vitro study using human primary chondrocytes showed that NAPA (2-(N-acetyl)-l-phenylalanylamido-2-deoxy-β-d-glucose), a derivative of glucosamine, inhibited IKKα kinase activity and its nuclear translocation.66 However, it remains to be demonstrated that NAPA does not also inhibit the nuclear translocation of specific transcription factors required for chondrocyte differentiation or other factors involved in collagen synthesis or remodelling. Importantly, there is no in vivo study linking NF-κB and IKKs to OA. Therefore, we are currently working to validate our in vitro observations in inductive OA disease models in mice to elucidate the mechanisms of action by which IKKβ and IKKα differentially drive pathways that impact on chondrocyte homeostasis to contribute to OA disease onset and/or progression.
References
Footnotes
KBM and MBG contributed equally to this work.
Funding Research related to this topic is supported by National Institutes of Health grants R01-AG022021 and RC4-AR060546 and by the CARISBO Foundation of Bologna (Italy); POS-FESR 2007–2013, Emilia Romagna Region and a scholarship from the Osteoarthritis Research Society International
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.
Data sharing statement No additional data are available.