Article Text

Abstract
Objective We have shown in vitro and in vivo that osteoclast maturation requires calcium-release activated calcium (CRAC) channels. In inflammatory arthritis, osteoclasts mediate severe and debilitating bone erosion. In the current study, we assess the value of CRAC channels as a therapeutic target to suppress bone erosion in acute inflammatory arthritis.
Methods Collagen-induced arthritis (CIA) was induced in mice. The CRAC channel inhibitor 3,4-dichloropropionaniline (DCPA) and a placebo was administered 1 day prior to collagen II booster to induce arthritis. Effects on swelling, inflammatory cell invasion in joints, serum cytokines and bone erosion were measured.
Results Assays, by blinded observers, of arthritis severity showed that DCPA, 21 mg/kg/day, suppressed arthritis development over 3 weeks. Bone and cartilage damage in sections of animal feet was reduced approximately 50%; overall swelling of joints was reduced by a similar amount. Effects on bone density by µCT showed clear separation in DCPA-treated CIA animals from CIA without treatment, while differences between controls without CIA and CIA treated with DCPA differed by small amounts and in most cases were not statistically different. Response was not related to anticollagen titres. There were no adverse effects in the treated group on animal weight or activity, consistent with low toxicity. The effect was maximal 12–17 days after collagen booster, during the rapid appearance of arthritis in untreated CIA. At 20 days after treatment (day 40), differences in arthritis score were reduced and tumour necrosis factor α, interleukin (IL)-1, or IL-6 in the serum of the animals were similar in treated and untreated animals.
Conclusions DCPA, a novel inhibitor of CRAC channels, suppresses bone erosion associated with acute arthritis in mice and might represent a new treatment modality for acute arthrits.
- Arthritis
- Bone Mineral Density
- Cytokines
- Treatment
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is already known about this subject?
3,4-dichloropropionaniline (DCPA) suppresses arthritis development in the collagen-induced arthritis (CIA) model.
DCPA reduced bone and cartilage damage as well as joint swelling by approximately 50% in CIA animals.
DCPA inhibited bone density reduction typically noted in CIA animals.
DCPA could provide a cost-effective, low toxicity treatment for arthritis associated bone erosion in humans.
Rheumatoid arthritis (RA) has a chronic relapsing course with bouts of acute arthritis leading to destruction often of multiple joints and usually requiring lifelong therapy. While there are infectious causes of inflammatory arthritis and genetic predilections, typically RA also is idiopathic. Treatments for inflammatory arthritis include antimetabolites, steroids and tumour necrosis factor α (TNF-α)-blocking molecules all of which can cause dangerous or debilitating side effects.1
We arrived at calcium-release activated calcium current (CRAC) inhibitors as a potential new means to treat acute arthritis through a series of studies of the role of calcium in osteoclast development. We found that the IP3R receptor calcium channel was necessary for osteoclast adhesion and activity,2 while others showed that IP3R-mediated release of calcium was required for activation of nuclear factor of activated T cells-1 (NFATc1)—a critical signal for osteoclast formation.3 We then demonstrated that osteoclastogenesis was dependent on CRAC channels activated by release of calcium stores: suppression of CRAC via siRNA or inhibition by the CRAC antagonist, 3,4-dichloropropionaniline (DCPA) impaired differentiation of bone degrading osteoclasts.4 Further, CRAC is required for normal T-cell differentiation, also via NFATc1 activation.5 Thus a web of lymphocyte and macrophage differentiation events depend on CRAC, all of which might be involved in arthritis progression.
While total developmental suppression of CRAC causes severe bone deformities and skeletal defects.6 In adults, where skeletal development is mature, short-term treatment is unlikely to cause major skeletal effects. However, in long-term treatment immunological effects are likely. Moreover, since the Th17 response important to the pathogenesis of RA7 appears to be dependent on the CRAC channel Orai18 ,9 we reasoned that partial CRAC inhibition by DCPA might effectively blunt acute arthritis. Although several CRAC inhibitors are known, we pursued DCPA in part because of very low toxicity; exposure of humans to DCPA in mg/kg quantities has minimal apparent toxicity,10 although massive doses, approximately 4–5 g/kg, can cause fatal methemoglobinaemia.11
We performed a trial of suppression of acute arthritis using controlled-release DCPA, at 10.5 or 21 mg/kg/day, in mice with acute collagen-induced arthritis (CIA). DCPA caused a dramatic suppression of bone destruction and of articular inflammation at the higher dose. The studies used blinded observation of joint swelling over 5 weeks, and histological and μCT examination of sections of bone in the feet. In addition, we studied anticollagen antibodies and inflammatory cytokines, which did not correlate with arthritis response at the times studied. Osteoclast and T-cell numbers were also examined by immunohistochemistry in affected joints and while osteoclasts numbers were reduced, T-cells were at similar density in inflammatory infiltrates in all groups.
Materials and methods
Animal studies
Six-week old male DBA/1 mice were purchased from Harlan Laboratories (Frederick MD) and housed in the WVU vivarium. These animals were acclimated at least 1 week prior to any experimental use. All animal procedures were approved by the WVU Institutional Animal Care and Use Committee.
Induction and assessment of arthritis
CIA was induced as described by Clutter et al.12 Briefly, bovine type II collagen (CII)(Elastin Products Co, Owensville, Missouri, USA) was emulsified in complete Freund's adjuvant consisting of 10 mL of Freund's Incomplete Adjuvant (Difco Laboratories, Detroit, Michigan, USA) plus 20 mg of Mycobacterium tuberculosis H37RA (Difco Laboratories). The CII (100 µg per animal; approximately 4 µg/kg) was injected intradermally on day 1 and 21 days later, a booster dose of 100 µg CII in Freund's incomplete adjuvant (Difco Laboratories) was administered. Inflammation was apparent 4–8 days after the second dose, in 80% of treated joints. At day 20 after primary immunisation, time-release pellets (Innovative Research of America, Sarasota FL) containing DCPA or the placebo, calibrated to release the stated doses for 21 days, were placed subcutaneously. Power analysis indicated that at least eight animals per CIA group were required to provide a valid statistical sample. Since induction of CIA does not occur in 100% of the treated mice, 12 mice in each CIA-induction group were initially started in the experiment. Treatment doses included 0 mg/kg (placebo), 10.5 mg/kg/day of DCPA or 21 mg/kg/day of DCPA were compared. Four untreated controls, that is, no CIA or DCPA treatment, were also included.
Mice were monitored for arthritis and scored in a blinded manner as described by Clutter et al.12 Briefly, swelling of paws was be graded on scale from 0 to 4 indicating number of swollen digits. All paws were evaluated, so that the maximal arthritic index per mouse was 16. Additionally, hind paw swelling was measured using digital calipers on day 0, and each day on days 23–40. Analysis of the bones and joints for arthritis was performed on H&E stained sections of hind paws, by blinded observation. This scored synovial expansion and inflammation, joint damage including pannus and bone degradation, each on a scale of 0–3, with maximum score of 9. For histological analysis, two paws from each animal were analysed separately and blindly, and are calculated as two specimens per animal.
Serum analysis for antibodies and cytokines
Heart blood collected at the time of euthanasia on day 40 was used for analysis. Plasma was separated by centrifugation and frozen in aliquots at −20°C until used. Production of anti-CII antibodies was evaluated by ELISA (Rheumera, Astarte Biologics, Redmond, Washington, USA) and cytokine concentrations were measured using V–PLEX panels (Meso Scale Discovery, Rockville, Maryland, USA) using the methods prescribed by the respective manufacturers.
Antibody labelling of sections
Histological sections from the feet of animals euthanised at 40 days, were stained using standard immunohistochemical methods to measure the effect of DCPA on osteoclast bone interface and T-cell density. Osteoclast bone interface density was determined by anti-ATPa3 (TCIRG) labelling, and the effect on CD3 T-cell density was determined using anti-CD3 labelling. Anti-TCIRG1 quantification was mouse monoclonal (clone 6H3) antibody (Sigma-Aldrich) at 1:100 dilution and CD3 quantification used mouse monoclonal antibody anti-CD3 PC3/188A (raised against amino acids 156–168 of the cytoplasmic domain of human CD3-ε) at a 1:100 dilution. Briefly, sections were blocked in phosphate-buffered saline (PBS) with 2% hydrogen peroxide for 5 min, then in PBS with 2% bovine serum albumin (BSA) for 2 h. The sections were incubated overnight with antibodies at indicated concentrations in PBS with 0.01% tween 20. After washing, sections were incubated for 1 h with biotinylated antimouse antibodies at 1:1000 dilution, washed again and incubated with streptavidin-horseradish peroxidase and diaminobenzidine substrate for 5 min. H&E counterstaining was performed to show tissue features. Imaging used a Nikon TE2000 inverted microscope, with 14-bit 2048×2048 pixel monochrome CCD camera and RGB filters to reconstruct colour (Spot, Sterling Heights, Michigan, USA).
μCT and morphometry
Analysis by μCT was as described.13 In brief, paws were scanned on a Viva CT40 instrument (Scanco, Bassersdorf, Switzerland) with three-dimensional (3D) reconstruction by the manufacturer's instrument-specific software. The scan section increment was 20 μm; 3D reconstruction used a density cut-off of 211 mg/cm3. Analysis of trabecular thickness and trabecular spacing used the plate model.
Statistics
Continuous data obtained at one time point (day 40), such as anticollagen antibody titres, bone density, bone volume, concentration of cytokines, osteoclast area and per cent area of periosteal T cells, were analysed using analysis of variance (ANOVA) followed by Dunnett's multiple comparison procedure (MCP) when the goal was to compare the CIA-treated groups to untreated controls. In addition, Tukey-Kramer analysis with adjustment for MCP and Fisher's LSD was performed for continuous variables when the goal was to compare the differences among the CIA-treated groups. The hind paw swelling data, recorded over time (day 0 and 23–40), were analysed by the repeated measures ANOVA, using the autoregressive autocorrelation structure and followed by Tukey-Kramer analysis with adjustment for MCP. Data for anticollagen antibody titres and paw swelling were Ln-transformed and reciprocal square-transformed, respectively, before ANOVA due to their positive skewness. Data of categorical or ordinal nature, such as arthritis index, were analysed by non-parametric Kruskal-Wallis test for each day. In addition, arthritis index data were grouped into two categories based on the response, 0 for arthritis index equal to zero and 1 for all mice with arthritic index greater than 0. These data were also analysed by Kruskal-Wallis test for each day, followed by the non-parametric Wilcoxon rank sum multiple comparison. A 60% power of statistical test was based on the average arthritic index for each group and the expected SD. Data were analysed using JMP and SAS software (SAS Institute Inc, Cary, North Carolina, USA). Significance criterion α for all tests was 0.05.
Results
In vivo analysis of CIA and the effect of DCPA treatment by continuous-release subcutaneous pellets. Mice showed no changes in body weight (data not shown), or increased lethargy or overt stress in any group, although severe inflammation of paws was induced by the CIA protocol. As measured by the arthritic index scoring each paw as 0–4 digits affected, on days 23–40 (after booster injections at day 20), 80% of mice responded to collagen injections by induction of arthritis (figure 1A). The collagen-treated animals receiving placebo (0 mg/kg/day DCPA) differed significantly from untreated controls, as expected (p<0.05 by Kruskal-Wallis test). The effect of 10.5 mg/kg/day of DCPA was modest and at most time points the results did not differ significantly by ANOVA from those of CIA animals treated with placebo (0 mg/kg/day DCPA). On the other hand, a dose of 21 mg/kg/day of DCPA reduced the arthritic index, strongly before day 37 and 20–50% at days 38–40. The arthritic index for animals treated with 21 mg/kg/day was statistically lower from that of CIA animals treated with placebo (Wilcoxon mean scores 14.5 vs 21, respectively, p<0.05 by Wilcoxon test, scores not shown). A scatter plot of the median arthritic scores of all animals on day 37 (day of maximum effect) is shown in figure 1B. In the CIA mice treated with 0 or 10.5 mg/kg/day DCPA >50% of the animals showed arthritis scores that variability was high. The majority of animals treated in the CIA animals treated with 21 mg/kg/day DCPA exhibited minimal arthritis scores, although three animals showed significant arthritis development. Hind paw thickness (figure 1C) was also measured with the average thickness for the untreated controls value of 2.29±0.03 mm. There were significant effect of treatment (p=0.02) and time (p<0.01) (data not shown) on paw thickness. Comparisons showed that on average, CIA-treated, low-dose DCPA (10.5 mg/kg/day) as well as placebo DCPA (0 mg/kg/day)-treated mice had significantly thicker paws (2.56±0.04 mm), than the untreated control mice (p<0.05 for each comparison). Those CIA-treated animals treated with 21 mg/kg/day DCPA had a paw thickness of 2.43±.04 mm which was not significantly different from the untreated controls (p=0.52). There was a relatively high variability in the data which likely reflects the all-or-none nature of arthritis; in some of the few mice treated with 21 mg/kg/day DCPA that still developed arthritis, it was severe.

The effect of 3,4-dichloropropionaniline (DCPA) treatment by continuous-release subcutaneous pellets on collagen-induced arthritis (CIA) as measured by the arthritic index. The data were gathered by a blinded observer. n=4 for untreated controls; in the CIA animals n=11 for animals treated with 0 mg/kg/day DCPA; n=10 for animals treated with 10.5 mg/kg/day DCPA; n=8 for animals treated with 21 mg/kg/day. (A) Median effect relative to CIA animals treated with 0 mg/kg/day DCPA (blue; open circles) and untreated controls (red; closed square). There is a trend toward efficacy at intermediate times in CIA animals treated with 10.5 mg/kg/day DCPA (green; closed circles) but only the CIA animals treated with 21 mg/kg/day DCPA (yellow; closed triangles) had a durable effect. (B) Scatter plot showing the arthritis scores at day 37. This demonstrates that >50% animals in the CIA animals treated with 0 mg/kg/day (blue; open circles) and 10.5 mg/kg/day DCPA (green; closed circles) show arthritis scores that exceed the mean and several animals showed the maximum arthritis score, whereas a majority of the animals /21 mg/kg/day DCPA (yellow; closed triangles) showed no or minimal arthritis scores. (C) Paw thickness, mean±SEM, for 4 control, 11 untreated CIA, and 8 high-dose DCPA/CIA animals. The difference between control and CIA animals is significant (*, p<0.05); the high-dose DCPA animals are not different relative to the other groups (ns).
Analysis of CIA in sections of feet
Hind paws of animals were fixed, decalcified, paraffin embedded, cut and stained with H&E (figure 2A–C). Arthritis was scored by a blinded observer on a scale of 0–9, evaluating two paws per animal. Sections were scored 0–3 for inflammation in periarticular tissue with 3 being suppurative inflammation, 0–3 for involvement of cartilage and the joint space, with 3 requiring a pannus and 0–3 for degradation of periarticular bone, with 3 being full thickness destruction of the end plate. Representative examples of paws from an untreated animal showing no inflammation (figure 2A), a DCPA-treated CIA animal showing measurable but not severe arthritis (figure 2C), and a placebo-treated CIA animal with severe disease (figure 2B) are shown. The quantification of arthritis severity by a blinded observer is shown in figure 2D. The high-dose DCPA/CIA group was statistically different from the CIA/placebo group with p=0.024. The untreated group was statistically different from all other groups with p<0.01. Results for the low dose (10.5 mg/kg/day) treatment group were poorly separated from results for other CIA groups (the low-dose DCPA-treated animals were therefore omitted from further analyses). Figure 2E shows the anti-type II collagen (CII) titres. Mean anti-CII titres in the 0 mg/kg/day DCPA group were 7.1±2.1×103, 5.1±1.1×103 in the 10.5 mg/kg/day DCPA group and 8.3±2.8×103 in the 21 mg/kg/day DCPA group; no significant differences among the CIA groups were found for anti-CII titres.

The effect of 3,4-dichloropropionaniline (DCPA) treatment on collagen-induced arthritis (CIA) at 40 days. (A–C) The top frame shows an ankle bone in an untreated animal (untreated control, histological score 0). The middle frame shows severe arthritis in CIA treated with 0 mg/kg/day DCPA, with severe synovial inflammation, pannus in the joint space and severe bone degradation (histological damage score 8). The bottom frame shows the results from CIA animals treated with 21 mg/kg/day; there is inflammation, with synovial thickening, but it is not severe (histological damage score 4). All fields are 1.2 mm across. (D) Severity of arthritis in each group scored by a blinded observer based on sections as in A–C. The histological joint damage index (HJDI) in CIA animals treated with 21 mg/kg/day DCPA (yellow bar) is statistically different from those treated with 0 mg/kg/day (blue bar), p=0.024. The untreated group was different from all other groups, p<0.01. The CIA animals treated with 10.5 mg/kg/day (green bar) were poorly separated on day 40 HJDI, and were not studied further. Both hind feet of each animal were individually processed and blindly scored, so the number of measurements is twice the number of animals and expressed as the mean±SEM HJDI. Animal numbers are as stated for figure 1. (E) Anti-type II collagen titre in CIA mice. The anti-type II collagen titre in CIA animals treated with 21 mg/kg/day DCPA (yellow bar) was 8.3 (±2.8)×103 and was not statistically different from those treated with 0 mg/kg/day (mean titre=7.1 (±2.1) × 103; blue bar). The mean anti-type II collagen titre of CIA animals treated with 10.5 mg/kg/day (green bar) was 5.1 (±1.1) × 103 and also was not statistically different from the other CIA animals. The anti-type II collagen titres in the untreated group were all below the level of detection. Values shown are the respective group mean±SEM.
μCT
There were clear erosions in CIA mice which were not observed in high-dose DCPA-treated animals (figure 3A–C), in accord with histology (figure 2A–C); high-dose CIA/DCPA could not be distinguished visually from untreated controls. Low-dose CIA/DCPA (10.5 mg/kg/day) was not studied. Quantitative morphometry in the paws, the most severely affected site in the skeleton, showed that bone density and bone volume per total volume was significantly decreased in CIA/placebo mice. The CIA/high-dose DCPA treatment group had density indistinguishable from that of untreated controls (figure 3D) and showed a trend toward greater density than in CIA/placebo mice (p=0.08). The bone volume/total volume for CIA/DCPA-treated mice did not differ from that of untreated controls, and it was statistically greater than for CIA/placebo animals (p=0.03). Other parameters of bone morphometry including trabecular thickness and spacing were not different between groups (data not shown). This reflects the fact that the damage to the bone in arthritis is specific to the joints, which are excluded when determining bone trabecular parameters.

μCT showing loss of bone in untreated animals and effect of high-dose 3,4-dichloropropionaniline (DCPA). (A–C) Examples of forelimb metacarpals in untreated mice, CIA mice treated with 0 mg/kg/day and CIA mice treated with 21 mg/kg/day DCPA after euthanasia at day 40. Arrows in (B) show cortical bone defects. (D–E) Bone density in total volume of bone and bone volume per total volume in four untreated controls and CIA animals treated with 0 (blue bar; n=9) or 21 (yellow bar; n=9) mg/kg/day DCPA each. For both measures the CIA animals treated with 0 mg/kg/day DCPA has significantly reduced bone; the CIA animals treated with 21 mg/kg/day DCPA is not different from the untreated control but is statistically greater than CIA animals treated with 0 mg/kg/day in BV/V.
Effect of CIA with and without DCPA treatment on serum inflammatory cytokines
We determined the effect of DCPA treatment on key serum cytokines including TNF-α, IL-1 and IL-6. The results showed strong induction of the cytokines by CIA but, surprisingly, showed no effect of DCPA on cytokine production despite strong suppression of arthritis by DCPA (figures 1⇑–3). Representative data from one of two separate analyses with essentially identical results is shown in figure 4. Interferon γ (IFN-γ), CXCL1, TNF-α, IL-1β and IL-6 (figure 4A–E) were all significantly increased in CIA/placebo animals compared to the untreated (no collagen injection) controls as expected. TNF-α was also significantly increased in the CIA/ high-dose DCPA mice compared to untreated controls. Comparison of CIA/placebo and CIA/high-dose DCPA groups revealed no significant reductions in any cytokines with CRAC inhibition. Based on these data, we hypothesise that the inhibitory effect of DCPA on osteoclasts was not secondary to changes in systemic cytokine levels, as these were not significantly affected. Nor did DCPA significantly affect production of IL-12p70 or IL-10, which also were measured (not shown). IL-17 was also assayed but systemic levels were too low for meaningful comparisons (see Discussion).

Effect of CIA and treatment with 3,4-dichloropropionaniline (DCPA) on production of key serum inflammatory factors. (A) IFN-γ. (B) CXCL1. (C) TNF-α. (D) IL-1β. (E) IL-6. One of two multiplex assays with similar results is shown. All of the cytokines shown were induced strongly with the exception of IL-10 (not shown). No cytokines differed between CIA animals treated with 0 (blue bar) or 21 mg/kg/day (yellow bar) DCPA. Statistical significance between groups is denoted by lower case letters above the bar. Mean levels not connected by the same letter are significantly different at p<0.05. IFN-γ, Interferon γ; IL-6, TNF-α, tumour necrosis factor α.
Effect of CIA and DCPA on osteoclasts and T cells in tissue sections
Antibody labelling for TCIRG1/ATPa3 was used to determine the effect of the DCPA treatment on osteoclasts in tissue (figure 5A–D). TCIRG1 is the large subunit of the osteoclast-membrane hydrogen pump; it is not expressed by macrophages (nor in other cells in the marrow); in contrast, enzymes including cathepsin K and TRAP, which are highly expressed by osteoclasts and often used as osteoclast markers, are also expressed in macrophages and so are less specific.14 ,15 TCIRG1 labelling revealed a large and statistically significant reduction in the bone surface area covered by osteoclasts in CIA/DCPA-treated mice compared to CIA/placebo mice (figure 5D). In contrast, anti-CD3 labelling showed the density of T cells in inflammatory infiltrates to be similar in the CIA/placebo and CIA/DCPA groups (figure 5E).
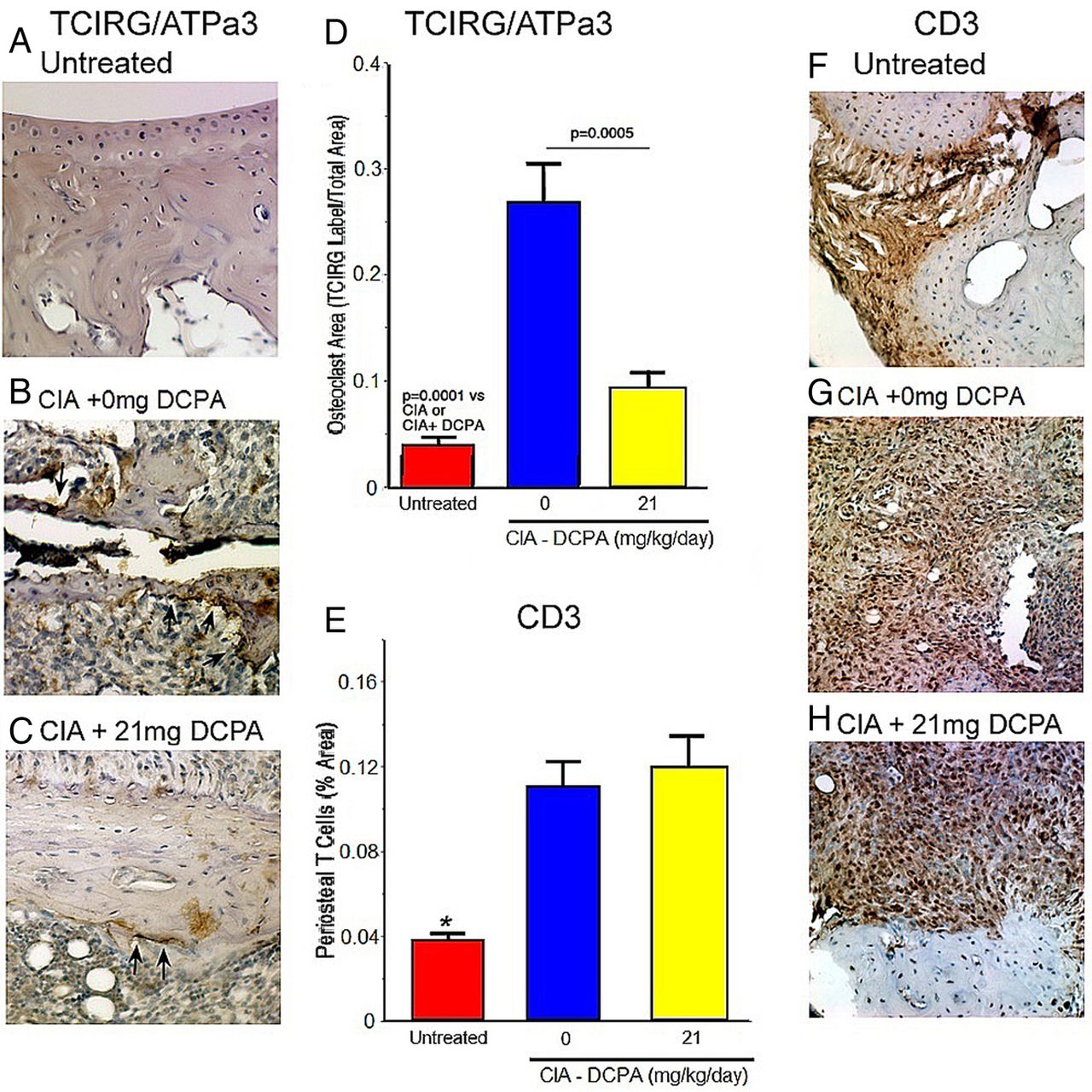
Effect of collagen-induced arthritis (CIA) and 3,4-dichloropropionaniline (DCPA) on osteoclast bone interface density determined by ATPa3 (TCIRG) labelling, and effect on CD3 T cells. (A–C) Typical section of each specimen type with ATPa3 antibody labelling. (D) Blindly scored area of ATPa3 labelling on bone. Mean fraction±SEM; n=5 for untreated and 9 each for CIA animals treated with 0 (blue bar) or 21 (yellow bar) mg/kg/day DCPA (right bar). All groups are statistically different with high confidence. (E) CIA animals have more T cells per section area, but T cell density in infiltrates is not reduced by DCPA treatment. The DCPA treatment greatly reduces inflammatory volume (figures 1 and 3) so total T cells will be far fewer. (F–H) Typical sections with CD3 antibody labelling. Note T cells in the ligaments of a no-CIA animal (arrow, F).
Discussion
We report an in vivo trial of a CRAC inhibitor on inflammatory arthritis using the mouse CIA model.12 The theoretical basis for the observed effect is that CRAC currents are essential to normal osteoclast development, and also are involved in T-cell activation which also promotes arthritis.4 ,5 Our results include a highly significant decrease in joint swelling, inflammatory infiltrate and bone damage. Despite the manufacturer's statement that DCPA release is constant, it is likely that the amount of released DCPA decreased during the past days, when arthritis effect diminished (figure 1A). These effects were independent from suppression of serum cytokines, suggesting that the approach may complement therapy that targets the inflammatory cytokines. The effects on many bone density and trabecular bone parameters showed uniformly that DCPA reduced the effects of CIA on bone health, although there was variability in statistical significance between individual parameters. On the other hand, an important unresolved issue is that it is likely, indeed almost certain, that at times with strongest arthritis suppression, effects on cytokine levels would occur, or if DCPA treatment were continued for longer times. In trabecular bone density, bone volume/total volume and trabecular spacing, the controls without were not statistically different from CIA treated with 21 mg/kg of DCPA, but CIA without treatment was well separated statistically from controls without CIA (figure 3). In further work, it will also be useful to compare the new modality of CIA treatment with other effective therapy as a positive control.
The pore-forming unit of the CRAC channel is Orai1,16 a four transmembrane-containing protein in the plasma membrane that is activated primarily by the endoplasmic-reticulum (ER) calcium sensor STIM1.17 Hence, in response to ER Ca2+ depletion, STIM1 translocates within the ER towards regions of close PM apposition where it interacts with Orai1.18 Using electrophysiological CRAC measurement and fluorescence analysis of STIM1/Orai1 association, we determined that DCPA inhibits CRAC channel activation by destabilising STIM1/Orai1 interaction, leading to CRAC channel closure.4 In that study, we further showed that DCPA inhibits osteoclast formation in vitro; we now show that that DCPA-induced CRAC channel inhibition leads to inhibition of osteoclast formation and function in vivo in an arthritis model.
Human Orai1 defects are now known, and are associated with defects including hypocalcaemia and muscular abnormalities.19 Although our mouse model showed no signs of serious metabolic or muscular anomaly, in larger animal studies it will be important to monitor these parameters. Oddly, some Orai1 polymorphisms also are associated with increased serum calcium and kidney disease, although the mechanisms are unclear.20 A dysfunctional STIM1 mutation, that is, in the calcium sensor required to activate Orai1 complexes, causes a syndrome with multiple developmental defects including anaemia, asplenia, myopathy and ichthyosis.21 Orai1 is also implicated in respiratory and neural development;22 ,23 in humans as in mice,6 complete absence of CRAC is probably lethal during early development or soon after birth. However, after normal development, at least for a time frame of 3–4 weeks, suppression of CRAC appears to be relatively benign.
Analysis of inflammatory cytokines showed that DCPA treatment at 21 mg/kg/day did not affect TNF-α, IL-1, IL-6, CXCL1, and IFN-γ (figure 4) all of which were upregulated 2.5-fold to >10-fold by the CIA. We did not detect significant IL-17; other work has shown that IL-17 after adjuvant-induced arthritis, at 35 days, had serum concentrations <1 ng/mL 35 days after arthritis induction, and which varied only approximately 20% between untreated animals and arthritic animals,24 so it is likely our assay was insufficiently sensitive to determine IL-17 effects. There is strong evidence for the involvement of IL-17 in cellular signalling involved in induction of arthritis,25–28 but it is not a major secreted serum protein; indeed, some amount of Orai1 signalling is believed to be required to support TH17 cell development.8 Typically, IL-17 production is assayed in tissue sections or cells, and further work will be needed to resolve the contribution of IL-17 suppression to the reduction of arthritis severity.
Recently, it was reported that another inhibitor of store-operated calcium entry, 4-methy-4’-[3,5-bis(trifluoromethyl)-1H-pyrazol-1-yl]-1,2,3-thiadiazole-5-carboxanilide, also called YM-58483 or BTP-2, at 10 mg/kg suppressed CIA and suppressed inflammatory cytokines IL-1, IL-6 and TNF-α to near-baseline levels at 10 days after induction.29 While it is expected that other inhibitors of CRAC channels would function similarly in suppressing arthritis, the differences in response of inflammatory cytokines are very interesting, although not unexpected. The inhibitor BTP2 suppresses cytokines including IL-2 and calcium influx by other calcium channels, specifically TRPM4, at low nanomolar concentrations.30 Overall, it is likely that BTP2 effects overlap those of DCPA, but that BTP2 has a range of effects that may be, at least in part, unrelated to CRAC suppression. It will be important to test additional time points to determine if DCPA has effects on some cytokines that were not resolved, but, importantly, at a time when skeletal effects were clearly suppressed by DCPA, IL-1, IL-6 and TNF-α were essentially identical in CIA with and without DCPA treatment (figure 4).
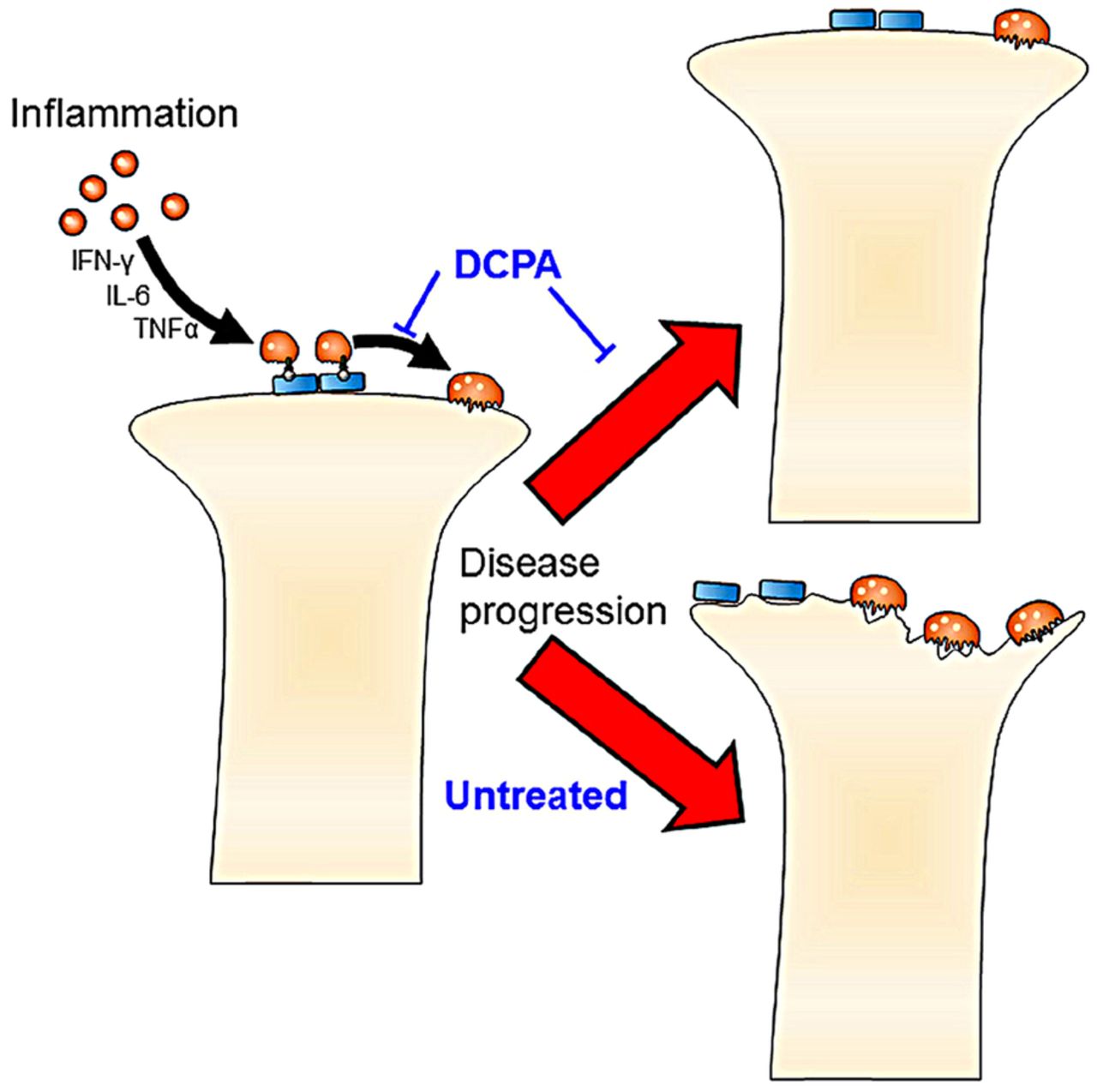
The clear effect of DCPA treatment on arthritis, despite no change in serum inflammatory factors, suggests that the direct blockade of CRAC activity by DCPA mediates the reduction in osteoclast maturation. This model (figure 6) will require further testing and refinement. However, the independence of the DCPA effect from suppression of the assayed serum inflammatory cytokines suggests that Orai1 antagonists may be valuable modalities for treating inflammatory arthritis, and possibly RA, by a mechanism independent of inflammatory cytokine suppression. However, serum cytokine levels may not reflect the levels of these cytokines within the joint spaces. It is likely that long-term strong inhibition of CRAC would result in severe immunological or bone effects, based on the knockout mouse phenotype,6 but treatment for several weeks had no effect on animal health, activity, or weight.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Model of the key functions of 3,4-dichloropropionaniline (DCPA) responsible for inhibiting collagen-induced acute arthritis. Independently of cytokines, the calcium-release activated calcium-inhibitor DCPA blocks maturation of osteoclasts, and probably also of T cells, contributing to arthritic destruction.
In conclusion, treatment of CIA with 21 mg/kg/day of DCPA dramatically suppressed bone erosion and bone loss by μCT that is associated with CIA, with no apparent effect on the general health of the animals or the increase in serum inflammatory growth factors characteristic of acute arthritis. This remarkable ability of a CRAC channel inhibitor to regulate bone erosion without interfering with immune function may lead to a valuable new tool for the treatment of inflammatory arthritis.
Acknowledgments
The authors thank Dr Johnny Huard, University of Pittsburgh, for assistance with μCT. Opinions expressed are not those of the Department of Veteran's Affairs. The authors also thank Kensey Bergdorf for her assistance with the figure illustrations.
References
Footnotes
Contributors HCB, JS, RH and JBB conceived and designed the experiments. HCB and JBB wrote the article with editorial contributions by others. IH, ME and JBB performed the CIA experiments. ILT, LJR, MRW and QCR performed in vitro analysis and histomorphometry. IH and LJR performed the statistical analysis on the data.
Funding Supported by the Department of Veteran's Affairs grants BX002490, NIH grants AR055208 to HCB, AR065407 to HCB, JS and LJR, AI073556 and AR056959 to RH, GM097335 to JS and ES011311 to JBB, and the WVU Clinical and Translational Science Institute and U54GM104942 to JBB and LJR.
Competing interests None declared.
Ethics approval The work was approved by the institutional animal care committees.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All useful data are included, either in the main paper or the supplemental information.