Article Text

Abstract
Objectives Neurological manifestations seem common in primary Sjögren's syndrome (pSS) but their reported prevalences vary. We investigated the prevalence and epidemiology of neurological manifestations in a French nationwide multicentre prospective cohort of patients with pSS, the Assessment of Systemic Signs and Evolution in Sjögren's syndrome (ASSESS) cohort.
Methods The ASSESS cohort, established in 2006, includes 395 patients fulfilling American–European Consensus Group criteria for pSS. Demographic and clinical data were compared between patient groups with and without neurological manifestations, and across patient groups with peripheral nervous system (PNS) manifestations, central nervous system (CNS) manifestations and no neurological manifestations.
Results Data at inclusion were available for 392 patients, whose mean age was 58±12 years. Mean follow-up was 33.9 months. Neurological manifestations were present in 74/392 (18.9%) patients, including 63 (16%) with PNS manifestations and 14 (3.6%) with CNS manifestations. Prevalences were 9.2% for pure sensory neuropathy, 5.3% for sensorimotor neuropathy, 1.3% for cerebral vasculitis and 1.0% for myelitis. Neurological manifestations were associated with greater pSS activity as assessed using the ESSDAI (9.4±6.8 vs 4.3±4.8; p<0.001) and proportion of patients taking immunomodulatory/immunosuppressive drugs (32.4% (24/74) versus 13.8% (44/318), p=0003). New neurological symptoms were more common in patients with than without prior neurological manifestations (RR=3.918 (95% CI 1.91 to 8.05); p<0.001).
Conclusions Prevalences of peripheral and central neurological manifestations in pSS are about 15% and 5%, respectively. Neurological manifestations are associated with greater pSS activity. New neurological manifestations are more common in patients with prior neurological involvement.
- Autoimmune Diseases
- Sjøgren's Syndrome
- Disease Activity
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Neurological manifestations seem common in primary Sjögren's syndrome (pSS) but their reported prevalences vary.
What does this study add?
Peripheral and central neurological manifestations in pSS are about 15% and 5%, respectively. These manifestations are associated with greater pSS activity and more common in patients with prior neurological involvement.
How might this impact on clinical practice?
Clinicians should be more attentive to neurological signs in pSS with high disease activity or prior neurological involvement.
Introduction
Primary Sjögren's syndrome (pSS) is an autoimmune disease characterised by xerophthalmia, xerostomia, arthralgia, myalgia and severe fatigue.1 Lymphocytic infiltration of the lachrymal and salivary glands is the hallmark of the disease and results in partial destruction of the gland parenchyma. Disease classification is based on the American-European Consensus Group (AECG) criteria or new preliminary American College of Rheumatology (ACR) criteria, which require a positive salivary gland biopsy or the presence of anti-Sjogren's syndrome A (anti-SSA) antibodies.2 Systemic manifestations of variable severity are common in pSS and play a major role in the prognosis.
The first studies of neurological manifestations in pSS were reported in the 1980s.3 Although neurological manifestations seem common, our recent review4 shows wide variations in reported prevalences. Nevertheless, the most recent data suggest a prevalence of about 20%. Neurological manifestations are the inaugural symptoms of pSS in a non-negligible proportion of cases. Thus, in a recent study, they occurred before the diagnosis of pSS in one-third of cases and were present at the time of diagnosis in two-thirds of cases.5 Obstacles to determining the exact prevalence of neurological manifestations in pSS include the limited size of available cohorts; differences in the inclusion criteria, particularly in retrospective studies, and patient recruitment modalities; and variations in the nature of the manifestations considered.6 Some studies focused on the peripheral nervous system (PNS)7 ,8 and others on the central nervous system (CNS),9 ,10 making the comparison of prevalences difficult.
Our primary objective was to study the prevalence of neurological manifestations in a French prospective cohort of patients with pSS in whom data were collected over 5 years. Secondary objectives were to investigate the demographic and clinical characteristics of patients with PNS, CNS or no neurological manifestations; the influence of pre-existing neurological manifestations on the development of new neurological manifestations; and pSS activity as assessed using the EULAR Sjögren's Syndrome Disease Activity Index (ESSDAI).11
Patients and methods
The study was sponsored by the Assistance Publique des Hôpitaux de Paris (AP-HP) and approved by the ethics committee of the Bichat Teaching Hospital (Paris, France) and French Data Protection Authority (CNIL) in 2006. All patients gave their informed written consent.
Patients
ASSESS (Assessment of Systemic Signs and Evolution in Sjögren's Syndrome) is a French nationwide multicentre prospective cohort established in 2006 with a grant from the French Ministry of Health (Programme Hospitalier de Recherche Clinique 2005 P060228). Its primary objective was to identify factors predicting systemic complications and lymphoma in pSS during a prospective 5-year follow-up.
Between 2006 and 2009, 395 consecutive patients fulfilling AECG criteria for pSS were included at 15 tertiary autoimmune disease centres. For each patient, all past and present systemic manifestations were recorded, including involvement of the skin, joints, lungs, kidneys, PNS, CNS, muscles and vessels. Cases of lymphoma were recorded, and the diagnosis and histological subtypes of previously diagnosed lymphomas were confirmed by having one of us (XM) review the medical and histological records. A standardised paper case report form (CRF) was completed prospectively by the participating clinicians each year at inclusion and then 1 year later. Systemic disease activity was assessed using the ESSDAI, a quantitative score that can range from 0 to 123 and covers 12 domains. The scoring system for PNS and CNS manifestations is shown in the appendix. The ESSDAI score was calculated retrospectively, by trained physicians, after validation of this tool. Once a year, each patient completed the EULAR Sjögren's Syndrome Patient Reported Index (ESSPRI)12 evaluating dryness, fatigue and pain. Dryness was assessed objectively at inclusion and 1 year later by determining the unstimulated salivary flow and performing Schirmer's test. The data were checked and then entered into an electronic database.
Neurological manifestations
We distinguished PNS and CNS manifestations. The PNS manifestations listed in the CRF were pure sensory neuropathy, sensorimotor neuropathy, neuronopathy, cranial nerve involvement, polyneuropathy, mononeuritis multiplex and polyradiculoneuropathy. The CRF also listed the following CNS manifestations: meningitis, meningoencephalitis, encephalitis, seizure, stroke, transverse myelitis and cerebral vasculitis. Past and present neurological manifestations were recorded separately. We analysed data at inclusion and during follow-up.
We also considered the following systemic manifestations, without separating them into subtypes: articular, cutaneous, muscular, renal, cardiovascular and pulmonary involvement; Raynaud's phenomenon; and vasculitis (including renal, cutaneous and neurological vasculitis). Given the difficulty in ascertaining that CNS manifestations are due to an autoimmune disease and not to another cause, we reviewed the medical files of patients with any of the above-listed CNS manifestations (except seizure) and/or cranial nerve involvement.
Statistical analysis
Continuous variables were described as mean±SD and compared using the Mann-Whitney test. Categorical variables were described as n (%) with the 95% CI and compared using Pearson's χ2 test. SPSS software V.21.0 (IBM, Armonk, New York, USA) was used for all analyses All significance tests were two-tailed, and values of p<0.05 were considered significant.
We analysed the baseline patient characteristics for which the relevant data were available. The prevalence of each neurological manifestation was computed by counting both past and present manifestations. We also performed a sensitivity analysis by calculating the prevalence of each neurological manifestation as described above, in the group of patients having available data both at baseline and after a 2-year follow-up. We compared patients with PNS and/or CNS manifestations, either past and/or present, to patients with no neurological manifestations at any time. The incidence of new neurological manifestations, defined as absent at baseline and present at any time during the first study year, was computed on the basis of all patients for whom the relevant data were available. We used Fisher's exact test to compare, in patients with available data at inclusion and after 2 years, incidence over 2 years between groups with and without neurological manifestations before the study and/or at baseline. Values of the ESSDAI computed using all items and without the PNS and CNS items were compared between patients with and without neurological manifestations (as defined above). Patients with systemic involvement were identified as patients whose ESSDAI was >0 and whose points were not related only to the presence of biological abnormalities. We then computed the mean ESSDAI domain subscores in patients with and without neurological involvement.
Results
Study patients
Of the 395 patients included in the ASSESS cohort, 392 had baseline data available, including 359 with data on neurological manifestations during the first year. Of the remaining 33 patients (8.4%), 27 had no clinical data at all and 6 no neurological data about the first year; past neurological manifestations were present in 5 and absent in 28. Figure 1 is the patient flow chart. Mean follow-up was 33.9 (±19.9) months for the 392 patients.

Patient flow chart (CNS, central nervous system; M, month; NS, nervous system; PNS, peripheral nervous system).
Demographic and clinical characteristics depending on baseline neurological profile
Table 1 lists the main characteristics in the four groups defined on the basis of the baseline neurological profile: any neurological involvement, PNS involvement, CNS involvement or no neurological involvement. Of the 392 patients, 367 (94%) were women and 25 (6%) were men. Mean age at diagnosis was 51±12 years (range, 17–82). The mean ESSDAI was 5.3±5.6 (range, 0–31). Compared to the group without neurological manifestations, the group with neurological manifestations had significantly greater disease activity (p<0.001) and significantly higher proportions of patients taking glucocorticoids (p<0.001), immunosuppressive/immunomodulating drugs (p<0.001), or rituximab (p=0.004) and/or vasculitis (p<0.001). Positive testing for anti-SSA and anti-Sjogren's syndrome B (anti-SSB) antibodies were associated with neurological manifestations (p=0.012 and p=0.018, respectively). Data about treatments or comorbidities were not available for some patients. The two groups were not significantly different for age at symptom onset or age at pSS diagnosis. Of the 53 patients with cryoglobulinemia, 1 had monoclonal, 17 mixed, 26 polyclonal and 9 undefined cryoglobulinemia.
Characteristics of patients with pSS included in the ASSESS cohort
Prevalence of peripheral and central neurological manifestations
At baseline, 74/392 (18.9%) patients had past or present neurological manifestations, including 63/392 (16.1%) with PNS involvement and 14/392 (3.6%) with CNS involvement. Table 2 reports the prevalence of PNS before and at the baseline visit. The most common PNS manifestation was pure sensory neuropathy (n=36, 9.2%), followed by sensorimotor neuropathy (n=21; 5.3%). Neuronopathy (ganglionopathy) was present in only 2 (0.6%) patients. Among patients with PNS manifestations, 53 had only one type (84.1%), 9 two types (14.3%) and 1 three types (1.6%). In the sensitivity analysis, in the group of patients having available data both at baseline and after a 2-year follow-up, prevalence was 19.2% for neurological manifestations (69/359), including 16.4% for PNS manifestations (59/359), 3.6% for CNS manifestations (13/359), 9.7% for pure sensory neuropathy (35/359), 5.3% for sensorimotor neuropathy (19/359), 0.6% for neuronopathy (2/359), 1.1% for cranial nerve involvement (4/359), 1.4% for polyneuropathy (5/359), 1.1% for mononeuritis multiplex (4/359) and 0.3% for polyradiculoneuropathy (1/359).
Peripheral nervous system involvement before and at inclusion
Central nervous system manifestations and cranial nerve involvement
CNS manifestations were recorded in 14 patients, including 1 with meningitis, 5 with seizures, 5 with cerebral vasculitis and 3 with transverse myelitis. Table 3 describes the patients with CNS manifestations other than seizures and cranial nerves involvement.
Features in the 14 patients with central nervous system and/or cranial nerve involvement
The medical file review of these patients showed that the most common therapeutic approach for severe CNS manifestations (that is, cerebral vasculitis and transverse myelitis) was combined glucocorticoid and cyclophosphamide therapy. Complete resolution of the severe neurological abnormalities was rarely achieved and improvements were obtained only inconsistently. Rituximab was used in 1 patient, who received four injections according to the usual schedule for vasculitis. MRI of the brain or spinal cord was performed in all patients. In patients with cerebral vasculitis, cerebral MRI showed extensive high-intensity signals in the white matter. Patients with transverse myelitis did not consistently have high-intensity signals by spinal-cord MRI; when this sign was absent, the diagnosis relied on the combination of typical clinical and electrodiagnostic features. As expected, cranial nerve involvement usually affected the Vth cranial nerve and was managed using standard pharmacotherapy.
New neurological manifestations: incidence and association with prior neurological involvement
During the first year of follow-up in the ASSESS cohort, 13 patients experienced new neurological manifestations, including 8 who developed pure sensory neuropathy, 3 neuronopathy, 1 sensorimotor neuropathy, 3 cranial nerve involvement, 1 encephalitis and 1 other central nervous system manifestation. Three patients had more than one manifestation. The annual incidence was 3.6% per year. During the second, third and fourth years of follow-up, respectively, 12, 6 and 4 patients experienced new neurological manifestations. Of the 35 patients, 16 did, and 19 did not, have a history of prior neurological involvement. Data about neurological manifestations during the first 2 years were available for 242 patients (37 with and 195 without neurological manifestations). Of the 25 patients in whom neurological manifestations appeared during the first 2 years of follow-up, 12 did and 13 did not, have a history of prior neurological involvement. New neurological manifestations were significantly more common among patients with than without prior neurological involvement: 32.4% (12/37) versus 6.7% (13/195) (RR=3.918 (95% CI 1.91 to 8.05); p<0.001).
ESSDAI values, neurological involvement and systemic involvement
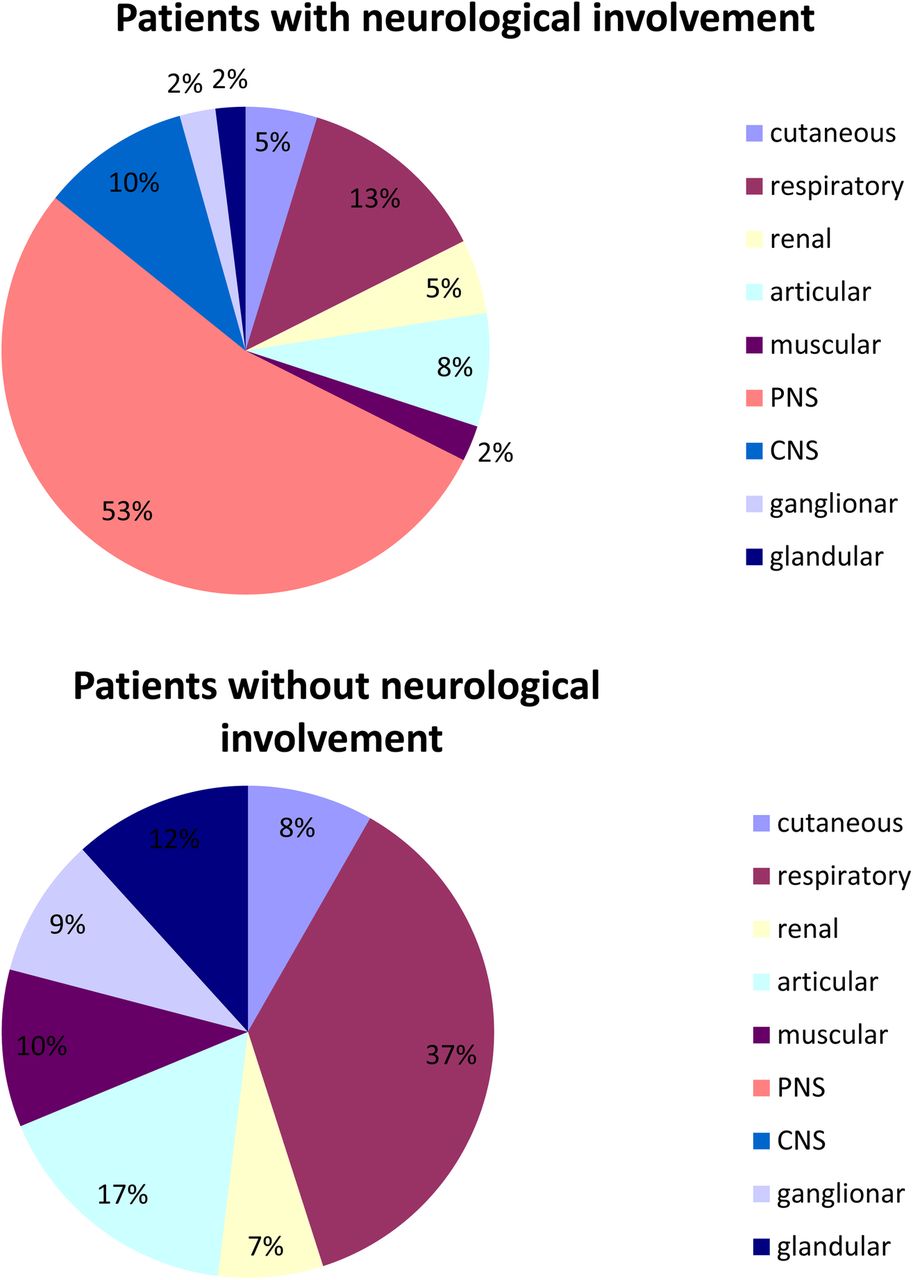
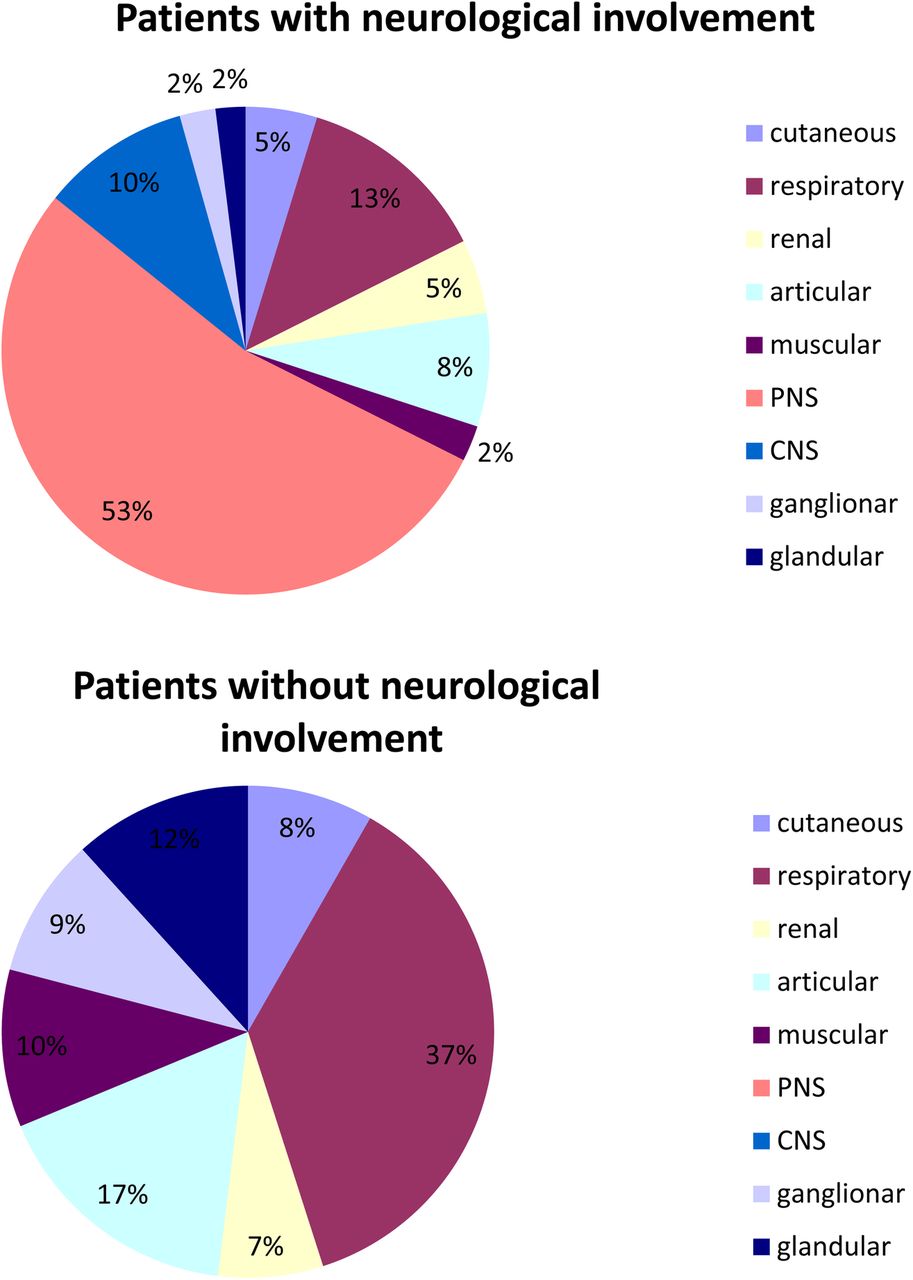
PNS manifestations contributed to the ESSDAI value in 35/379 (9.2%) patients including 1 with high, 18 with moderate, and 16 with low disease activity. CNS manifestations contributed to the ESSDAI value in 8 (2.1%) patients including 2 with high and 6 with moderate disease activity. On average, patients with and without neurological manifestations had respectively 1.8±1.2 and 1.2±1.1 ESSDAI domain affected (p<0.0001). The mean ESSDAI in patients with and without neurological involvement was 9.4±6.8 and 4.3±4.8, respectively (p<0.001). This ESSDAI difference was no longer significant after excluding the points contributed by PNS and CNS manifestations (4.9±4.4 and 4.3±4.8, respectively). Figure 2 showed the distribution of points across the ESSDAI domains in patients with neurological manifestations and in those with systemic involvement but no neurological manifestations. In the patients with neurological manifestations, slightly less than half the points were contributed by non-neurological systemic manifestations. In the patients without neurological manifestations, respiratory and articular involvement accounted for most of the systemic manifestations.
{kind=link}
{kind=link}
Distribution of points (%) across the ESSDAI domains in patients with neurological involvement and in those with non-neurological systemic involvement. CNS, central nervous system; ESSDAI, EULAR Sjögren's Syndrome Disease Activity Index; PNS, peripheral nervous system.
Discussion
The prevalence of neurological manifestations in a French cohort of 392 patients with pSS was 18.9%. The PNS was involved far more often than the CNS (16.1% and 3.6% of patients, respectively). These findings are consistent with previous reports indicating that about 20% of patients with pSS have neurological manifestations. Nevertheless, few studies were as large as our study. A 2013 Spanish study of systemic involvement in 921 patients with pSS found PNS and CNS manifestations during follow-up in 10.4% and 2.7% of patients, respectively.13 The apparent discrepancy between these findings and ours may be ascribable to the use in the Spanish study of the ESSDAI to define neurological involvement, as opposed to a specific list of neurological manifestations in the CRF used in our study. Past or subtle neurological manifestations may therefore have been underestimated in the earlier study.13 On the other hand, limitations of our data collection method include the need for good recall about past manifestations and the limited number of manifestations listed in the CRF (7 PNS and 7 CNS manifestations). Neuronopathy seems rare, as only two patients had this abnormality at baseline. However, during the first year of follow-up, neuronopathy was diagnosed in three additional patients. These new diagnoses may be ascribable to the close patient follow-up and absence of a need for imaging or histological proof to diagnose neuronopathy according to the criteria in the CRF. Two of those patients underwent an electromyography.
CNS manifestations in pSS are receiving increasing research attention. However, the type of CNS manifestations considered varies widely. Thus, some studies focused on transverse myelitis14 or another single manifestation,15–18 whereas others considered a vast array of abnormalities ranging from meningoencephalitis to cognitive dysfunction.10 ,19 The neurological manifestations ascribable to, as opposed to concomitant with, pSS need to be better delineated. The CNS manifestations investigated in our study were cerebral vasculitis, seizures, stroke, transverse myelitis, meningitis, encephalitis and meningoencephalitis. No patient had encephalitis or meningoencephalitis, and only one had meningitis. Although this last patient had clinical and biological features strongly suggestive of lymphocytic meningitis and responded to immunosuppressive drug treatment, the causal link between the meningitis and the pSS remains debatable. Overall, in only a few of the 14 patients with CNS manifestations was a causal link to pSS supported by strong arguments (e.g, suggestive imaging findings or cryoglobulinemia). For example, our fifth patient, presenting urinary dysfunction, underwent an MRI which was not supportive. In those cases, diagnoses rely only on clinical aspects and partially in the evolution after immunosuppressive therapy. This underlines the complexity of establishing a causal link between central nervous system manifestations and pSS. Nevertheless, myelitis, cerebral vasculitis and pSS are sufficiently rare20 that the presence of either neurological manifestation and of pSS in the same patient probably indicates a causal link.
Two recent studies showed widely differing prevalences of CNS manifestations in patients with pSS, namely, 67.5% (81/120)10 and 5.8% (25/424).21 This difference is probably ascribable in part to the patient recruitment at a rheumatology department in the study with the low prevalence and a neurology/psychiatry department in the study with the high prevalence. Furthermore, the nature of the manifestations differed in the two studies. The rheumatologists21 considered motor and/or sensory deficits, aphasia, seizures, brainstem syndrome, cerebellar syndrome, acute or subacute encephalopathy, cognitive dysfunction, psychiatric abnormalities, myelitis, neurogenic bladder, lower motor neurone disease, Brown-Sequard syndrome, optic neuropathy and multiple sclerosis-like disease. In contrast, the neuropsychiatrists10 recorded neuropathy, neuropathic pain, myopathic pain, fibromyalgia, movement, seizures, cerebellar signs, sensory impairments, pyramidal signs, neuromyelitis optica, meningoencephalitis, fatigue, headache, psychiatric disorders and cognitive disorders. Both studies collected highly detailed information on multiple symptoms, whereas we focused on syndromes and therefore had fewer details about the clinical manifestations. Our results are similar to those reported in the study21 published by Massara et al which reported about 5% of patients having CNS abnormalities. The high prevalence in the neuropsychiatry study10 is probably ascribable to the broad definition of manifestations that are common in pSS (e.g, fatigue, headache).22–25
The ESSDAI is a validated tool26 ,27 for evaluating the activity of pSS28 and for predicting, together with immunological markers, the risk of death related to pSS.29 In our study, the mean ESSDAI was higher in the group with neurological manifestations, but there was no longer a significant difference when only non-neurological systemic manifestations were used to determine the ESSDAI. The neurological items in the ESSDAI do not cover the full range of neurological manifestations seen in pSS. Past neurological manifestations were considered in our prevalence study but, when fully resolved, did not contribute to the ESSDAI value. For example, patient number three had a history of cranial nerve involvement successfully treated with gabapentin, which contributed no points to the ESSDAI. The same occurred for patient number 12, who was symptom free after discontinuation of a 2-year treatment for meningitis. In patients with neurological involvement, the ESSDAI values obtained without counting the points related to PNS and/or CNS manifestations were higher than those in patients without any systemic manifestations, suggesting an association between neurological and other systemic manifestations of pSS. Similar associations have been observed among other systemic manifestations, such as muscular, renal, cutaneous, respiratory, glandular, articular and lymph node involvement.
Our study has several limitations. First, all the study centres were in France. However, cohorts from other parts of Europe had a similar prevalence of neurological manifestations of 15–20%. Second, neurological manifestations were defined on the basis of criteria in the CRF, given the absence of a clear definition of pSS-related neurological manifestations. The seven PNS and seven CNS manifestations described in the CRF covered a large part of the neurological manifestations seen in pSS. Nevertheless, a small proportion of such manifestations may have been overlooked. Finally, our chart review of patients with CNS or cranial nerve involvement provided interesting information on the nature and treatment of these abnormalities, but our study shares with others a too small number of patients presenting with CNS manifestations to allow firm conclusions.
Our study also has strong points, including the large number of patients (395), sufficient for the evaluation of the most common neurological manifestation and a good representativity given the distribution of the inclusion centres (15) throughout France. Most of the patients (359/395) had a follow-up longer than 1 year and more than half had information about neurological manifestations after a 2-year follow up. The agreement between our findings and those of smaller French cohort studies and large European studies supports the validity of our prevalence data. Thus, about 20% of patients with pSS experience neurological manifestations, 5% CNS manifestations and 15% PNS manifestations.
To conclude, neurological manifestations are common in pSS, with about 20% of patients being affected. PNS involvement is far more common than CNS involvement, although the latter can induce severe morbidity. Pure sensory and sensorimotor neuropathies were the most common PNS manifestations. The development of new neurological manifestations was more common among patients with prior neurological involvement.
References
Footnotes
Funding This work was supported by grants from the French Ministry of Health (Programme Hospitalier de Recherche Clinique) awarded in 2005 to the ASSESS national cohort and the French Society of Rheumatology.
Competing interests None declared.
Patient consent Obtained.
Ethics approval This study obtained an Ethics Committee/Institutional Review Board approval.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All data about neurological involvement in pSS are in the manuscript.