Article Text

Abstract
Objectives (1) Hypothesis testing of the potency of rituximab (RTX) in preventing fibrotic complications and (2) assessing acceptability and feasibility of RTX in early systemic sclerosis (SSc).
Methods A small, 24-month, randomised, double-blind, placebo-controlled, single-centre trial in patients with SSc diagnosed <2 years was conducted. Patients received RTX or placebo infusions at t=0, t=15 days and t=6 months. Patients were clinically evaluated every 3 months, with lung function tests and high-resolution CT every other visit. Skin biopsies were taken at baseline and month 3. Immunophenotyping of peripheral blood mononuclear cells was performed at every visit, except at months 9 and 18. Adverse events, course of skin and pulmonary involvement and B cell populations in skin and peripheral blood were evaluated.
Results In total 16, patients (rituximab n=8, placebo n=8) were included. Twelve patients had diffuse cutaneous SSc. Eighty-eight adverse events (RTX n=53, placebo n=35, p=0.22) and 11 serious adverse events (RTX n=7, placebo n=4, p=0.36) occurred. No unexpected RTX-related events were observed. Mean skin score over time did not differ between the groups. Over time, forced vital capacity and extent of lung involvement slightly improved with RTX, but this difference was insignificant. In peripheral blood B cells depletion was demonstrated.
Conclusions No unexpected safety issues were observed with RTX in early SSc. Although this small trial could not confirm or reject potential efficacy of RTX in these patients, future placebo-controlled trials are warranted, specifically in the subgroup of patients with pulmonary involvement.
Trial registration number EudraCT 2008-07180-16; Results.
- Systemic Sclerosis
- B Cells
- Treatment
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is already known about this subject?
In the tight-skin mouse model of systemic sclerosis, anti-CD20 therapy suppresses development of skin fibrosis in newborn tight-skin mice.
Open-label studies in patients with systemic sclerosis showed possible clinical benefit of rituximab on skin and lung involvement.
In systemic sclerosis patients, rituximab treatment results in depletion of CD20-positive B cells from peripheral blood and skin.
What does this study add?
This study reports data on the course of skin and lung involvement in patients with early systemic sclerosis patients that participated in a small randomised placebo-controlled phase II trial with rituximab.
Rituximab might be of benefit for pulmonary involvement in early systemic sclerosis.
Immunologic analyses demonstrate depletion of the different B cell subsets from peripheral blood; immunohistological analyses show that the number of B cells present in skin is low, and also after treatment with rituximab, CD79 positive cells remain.
How might this impact on clinical research?
A larger phase III trial of rituximab in patients with early systemic sclerosis and pulmonary involvement can be considered.
Introduction
Systemic sclerosis (SSc) is an autoimmune disease that is characterised by the triad of microvascular damage, dysregulation of innate and adaptive immunity and generalised fibrosis in multiple organs.1
The pathogenesis of SSc is poorly understood and treatment is organ and symptom based. Current therapy targeting the dysregulated immune system, supported by clinical trial data, includes methotrexate for early skin involvement,2 3 cyclophosphamide followed by mycophenolate mofetil or azathioprine for lung involvement4–8 and autologous haematopoietic stem cell transplantation for severe, diffuse cutaneous systemic sclerosis (dcSSc).9
Experimental data suggested a key role for B cells in the pathogenesis of SSc.10 B cells seem to overexpress the stimulatory receptor CD19 and interleukin-10-producing regulatory B cells are decreased.11
Previous observational open-label studies of anti-CD20 therapy (rituximab (RTX)) and a nested case–control study in SSc showed potential efficacy for skin disease and stabilisation of internal organ disease in dcSSc.12–21 Since natural disease course is variable and difficult to predict, these results are difficult to value and need to be replicated in randomised controlled trials (RCTs).22
We hypothesised that the window of opportunity for rituximab (RTX) in patients with SSc lies early in the disease course, when fibrotic complications are yet to develop. This hypothesis is based on observations in a study in mice in which B cell depletion with anti-CD20 was effective in prevention of skin fibrosis in newborn tight-skin mice while no benefit was observed in tight skin mice with established disease.23 Additionally, B cell activating factor (BAFF) levels in these mice were elevated at 4 weeks after birth, while normalised at week 12 when skin fibrosis was established.24
Based on these observations, we aimed to test the hypothesis that RTX can prevent development of severe fibrotic complications in early SSc. Additionally, safety and feasibility of RTX in early SSc is described, together with the influence of RTX on immune cell subsets in peripheral blood and in skin tissue in patients with SSc.
Methods
Trial design
The RTX in early SSc trial was designed as a 24-month, parallel, double-blind, placebo-controlled randomised trial. Randomisation was performed in a 1:1 ratio by the Pharmacy of the Leiden University Medical Center (LUMC), Leiden, The Netherlands. Ethical approval was obtained from the Medical Ethical Committee (METC) of the LUMC and patients gave written informed consent. The study was monitored by a Data and Safety Monitoring Board until completion.
Patients
Between June 2010 and February 2014, patients with an established diagnosis of SSc according to the American Rheumatology Association (ARA) criteria25 within the last 24 months before enrolment and aged between 18 and 70 years were included. Previous immunosuppressive therapy was allowed and continued throughout the trial. Patients with a history of deep tissue infection within 1 year prior to baseline, patients with chronic or recurrent infections and patients with a history of cancer were excluded.
Procedures
Patients received intravenous 1000 mg RTX (MabThera/Anti-CD 20 mAb) or placebo (0.9% NaCl) on day 1 and day 15 as induction treatment. Consolidation treatment consisted of a single intravenous treatment with 1000 mg RTX or placebo (0.9% NaCl) at 6 months. Each infusion of RTX was given together with intravenous methylprednisolone 100 mg, oral paracetamol 1000 mg and intravenous clemastine 2 mg. Placebo-treated patients received 1.6 mL 0.9% NaCl together with oral paracetamol 1000 mg and intravenous clemastine 2 mg. Concomitant medications or other treatments deemed necessary for patients’ supportive care and safety were allowed at the discretion of the treating physician. Patients, physicians and the observers performing the skin score were blinded for treatment allocation.
Data collection
Patients were seen every 3 months during the first year and every 6 months thereafter, for physical examination including the modified Rodnan skin score (mRSS), and assessing toxicity (National Cancer Institute, common toxicity parameters),26 urinalyses and laboratory testing (at t=0, 3, 6, 12 and 24 months also including samples for immunophenotyping of peripheral blood) for a total follow-up of 24 months. Skin scores were assessed by an experienced research nurse (AV) and a research physician who was trained by AV (JM). In two-thirds of cases, the skin score was assessed by AV and in one-third by JM. Patients filled out the following questionnaires at every visit: Short Form 36 (SF-36),27 28 EurolQol-5D (EQ-5D)29 30 and Health Assessment Questionnaire Disability Index (HAQ-DI).31 32 Lung function tests including forced vital capacity (FVC) and diffusing capacity of the lungs for carbon monoxide (DLCO), high-resolution CT (HRCT) of the thorax and echocardiography were performed every 6 months. HRCTs were assessed using Goh criteria evaluating the extent of lung involvement at five levels: (1) origin of great vessels; (2) main carina; (3) pulmonary venous confluence; (4) halfway between the third and fifth section; (5) immediately above the right diaphragm.33 Scoring was performed consensus based, by two observers (AS and LK).
For histological and immunohistochemical analysis of the skin, 4 mm skin biopsies were obtained from the dorsal side of the forearm, within 1 cm of each other, at baseline and at 3 months.
Immunohistochemistry of skin tissue was performed on 4 µm thick sections on polysine-coated slides. After routine deparaffinisation and rehydration, antigens were retrieved in a tissue microwave for 12 min at 98°C with a Target Retrieval Solution (Tris)/EDTA pH 9. Quenching of endogenous peroxidase activity was performed with 1% hydrogen peroxide in methanol for 10 min. Biopsies were incubated with: H&E staining (4085.9005 and 4085.9002; Klinipath; Duiven, Netherlands) (general histopathology assessment and mononuclear infiltration), PBS/1%BSA for 1 hour with CD3 (1.41 µg/mL; M7254; DAKO) (T-cells), CD68 (0.12 µg/mL; M0814; DAKO) (macrophages), CD79a (1.875 µg/mL; M7050; DAKO) (B cells including plasma cells). Human tonsil specimens were used as a positive control for antibodies.
Stained sections were coded and scored by three observers who were unaware of clinical data and treatment regimen (AD, KQ, MB) with respect to the following points: histological signs of scleroderma skin (such as presence and entrapment of adnexa), mononuclear infiltration (semiquantitative scale), T cell infiltration (semiquantitative scale), B cell infiltration (semiquantitative scale) and macrophage infiltration (semiquantitative scale). Semiquantitative scoring for lymphocyte and macrophages was based on the scoring scale for lymphocytes proposed by Roumm et al with ‘0’ being a few scattered cells, ‘1’ a maximum number of cells per collection of at least 10, ‘2’ a maximum number of cells per collection between 10 and 50 and ‘3’ a maximum number of cells per collection of at least 50.34 Median scores were used for analysis.
Peripheral blood mononuclear cells (PBMCs) were isolated from 50 mL of peripheral blood by Ficoll-Paque gradient centrifugation, incubated for 20 min at 4°C and subsequently stained with CD3 allophycocyanine (APC) (clone SK7), CD4 Fluorescein Isothiocyanate (FITC) (clone RPA-T4), CD8 phycoerythrin (PE) (clone RPA-T8), CD14 FITC (clone MSE2), CD16 PE (clone B73.1), CD19 PerCPcy5.5 and APC (clone Sj25C1), CD20 FITC (clone L27), CD27 PE (clone L128), CD38 PerCPcy5.5 (clone HIT2), CD56 PE (clone MY31), Polyclonal IgA FITC (DAKO), IgD FITC (clone IA6-2), IgE Alexa Fluor 488 (gift from University of Antwerp), IgG FITC (clone G18-145), IgM APC and FITC (clone G20-127, all (except IgA and IgE) BD Biosciences). For isotype controls, IgG1 APC and PerCPcy5.5 (clone MOPC-21), IgG1 FITC and PE (clone X40), IgG2a FITC (clone X39), IgG2b FITC (clone 27–35), rabbit immunoglobulin fraction (DAKO) and polyclonal swine anti-rabbit immunoglobulins FITC (DAKO), all (rabbit immunoglobulins) BD Biosciences, were used. In addition, B cells and plasmablasts were stained with CD20, IgA, IgD, IgE, IgG, IgM and appropriate isotype controls.
Enzyme-linked immunosorbent spot (ELISPOT) technique was used to detect functional antibody-secreting cells, with the use of goat antihuman IgG, IgA and IgM (Sanbio BV, Uden, The Netherlands) for coating (10 µg/mL in coating buffer, 100 µL/well) of ELISPOT plates (Millipore, The Netherlands). Plates were incubated overnight at 4°C, washed twice with PBS and blocked with 200 µL/well culture medium (Iscove's Modified Dulbecco's Medium(IMDM) +10% FCS+200 mM L-glutamine+100 µg/mL penicillin/streptavidin) for 2 hours at 37°C in a 5% CO2 atmosphere. PBMCs were titrated on the ELISPOT plates in duplicate wells, and the plates were next incubated at 37°C in a 5% CO2 atmosphere overnight. The following day cells were discarded and washed from the plates with PBS/0.05% Tween 20 and tap water. Spots were visualised by detection with alkaline phosphatase-conjugated goat antihuman IgG, IgM or IgA (Biosource, USA) followed by substrate 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium (Sigma-Aldrich, St Louis, MO, USA) at 100 µL/well. ELISPOTs were analysed using a stereomicroscope (Bioreader 5000; BIO-SYS GmbH, Karben, Germany).
Study end points
In the design of the trial, the following parameters were defined as major clinical end points: treatment-related mortality, toxicity and clinical efficacy of rituximab. Efficacy was defined as progression-free survival, with progression defined as any or a combination of the following changes relative to baseline at two consecutive evaluations: death, ≥10% drop in predicted FVC,33 ≥15% drop in predicted DLCO,33 ≥15% drop in left ventricular ejection fraction (LVEF), body weight,35 ≥30% drop in creatinine clearance,36 ≥30% increase in mRSS,37 38 ≥0.5 point increase in HAQ-DI.38 The secondary end points defined were changes in mRSS (minimally important difference 3.2–5.3),39 FVC, DLCO, HAQ-DI (minimally important difference 0.10–0.14),39 left ventricular ejection fraction, creatinine clearance, SF 36, EQ-5D, presence of interstitial lung disease (ILD) as reported by HRCT thorax and skin biopsy scores.
However, unfortunately, the trial had major recruitment problems. In a time span of nearly 4 years, 17 patients had been included in the trial. Based on this low inclusion rate, the METC advised to prematurely end inclusion and evaluate study outcome 1 year after inclusion of the last patient. As one patient showed early dropout, n=16 patients (n=8 RTX, and n=8 placebo) had data available for analysis. All data collected by 30 June 2015 were included in the analysis. Based on the small sample size, we chose to focus on presentation of changes in mRSS, FVC, DLCO and extent of ILD as represented by Goh scores. Adverse events and serious adverse events and changes in HAQ-DI, LVEF, creatinine clearance, SF-36, EQ-5D were assessed for both treatment groups. Immunologically, the influence of RTX on mononuclear cell subsets in PBMCs and skin tissue was evaluated and described as planned.
Statistical analysis
As all patients participating in the trial also participated in the care programme of the LUMC,40 including annual and comprehensive diagnostic evaluation with informed consent for use of data, missing data were imputed from clinical files when possible with a maximum time frame of 6 months between data collection and planned data collection according to the trial schedule. This way, skin scores were available up to 24 months for all patients, pulmonary function tests for n=13/16 of patients at t=12 months and n=7/16 at t=24 months. HRCT images were available for scoring in n=15/16 at t=12 months and n=7/16 at t=24 months. Peripheral blood samples for PBMC assessment were available in n=15/18 at t=12 months and n=11/18 at t=24 months.
Primary analyses included mean change over time in mRSS, percentage of predicted DLCO, percentage of predicted FVC, extent of ILD as represented by Goh scores and HAQ-DI, for both treatment groups. Additionally, mortality, treatment toxicity and efficacy according to prespecified criteria were evaluated for both groups.
Ninety-five per cent CIs were computed where appropriate, with p values less than 0.05 (two-sided) considered statistically significant. Binary variables were analysed by Fisher exact test.
To assess the influence on clinical efficacy analyses of patients included under protocol violation, analyses were repeated excluding these patients. Interobserver agreement of skin biopsy scoring was evaluated using Fleiss’ kappa.41 Statistical analyses were performed using IBM SPSS Statistics 23 and GraphPad Prism 6.
Results
Patients and treatment
From April 2010 to February 2014, 17 patients were included, of which nine patients were randomised to RTX and eight were randomised to placebo (figure 1). All patients included fulfilled ARA criteria as well as ACR/EULAR 2013 SSc criteria.42 Two patients were included with a time since diagnosis of >24 months: one RTX patient (time since diagnosis 3.5 years, time since non-Raynaud 3.5 years and time since Raynaud 5.3 years) and one placebo patient (time since diagnosis 4.2 years, time since non-Raynaud 4.2 years and time since Raynaud 4.9 years). One patient (placebo group) died due to disease progression, after dropout at 6 months because of active disease. One patient did not start the allocated treatment based on active, severe disease with rapid progression of skin score and myocarditis/pericarditis, for which the treating physician judged the chance for placebo as possibly life-threatening. Baseline characteristics of this patient did not differ from other patients included.

Flow of rituximab in early systemic sclerosis trial. aOne patient refused to have a treatment at 6 months. bTwo patients incidentally received a dose of verum methylprednisolone instead of placebo methylprednisolone together with the RTX/placebo infusion. RTX, rituximab; AE, adverse event; SAE serious adverse event.
Baseline characteristics of the 16 patients included for analysis (RTX n=8, placebo n=8) were similar between the two groups, although there was a minor difference in disease duration, with slightly longer disease duration in the placebo group (table 1). The median follow-up of patients was 19.1 months (IQR 17.6–24.4). According to Goh criteria, mean extent of lung involvement at baseline was 9.5%±11.0 for RTX and 6.9%±10.8 for placebo (p=0.65).
Baseline characteristics of study patients
Previous immunosuppressive therapy included prednisone (RTX n=2, placebo n=1), methotrexate (RTX n=3, placebo n=0) and azathioprine (RTX n=2, placebo n=0). At start of the trial, use of immunosuppressive medication included prednisone (RTX n=2, placebo n=0), methotrexate (RTX n=5, placebo n=3), plaquenil (RTX n=1, placebo n=1) and mycophenolate mofetil (RTX=1, placebo n=1). During the trial background immunosuppressive treatment was changed in two patients in the RTX group: methotrexate was stopped at the 18-month visit in both cases (n=1: pregnancy; n=1: pancytopenia).
Analysis of clinical disease parameters
Course of changes in mRSS, FVC, DLCO and HAQ are shown in figure 2. There were no significant differences in change between baseline and 12-month follow-up of mRSS (placebo −1.8 vs RTX −3.6, p=0.95), FVC (placebo +0.3 vs RTX +4.7, p=0.43), DLCO (placebo −0.3 vs +0.7, p=0.91) and HAQ (placebo +0.18 vs RTX 0.0, p=0.94). Also, at 24-month follow-up, there were no significant differences in change from baseline in mRSS (placebo −1.9 vs RTX −5.3, p=0.95), FVC (placebo −1.4 vs RTX+4, p=0.65), DLCO (placebo −2.2, RTX −6.0, p=0.77) and HAQ (placebo 0.2313 vs RTX −0.0675, p=0.94) results. Numerically, n=4/8 rituximab versus n=2/8 in placebo improved >5 points in mRSS; there were no improvers in either FVC or DLCO (minimal important difference 10%) and n=1/8 in RTX vs n=0/8 in placebo improved in HAQ (minimal important difference 0.5 points) after 1 year.
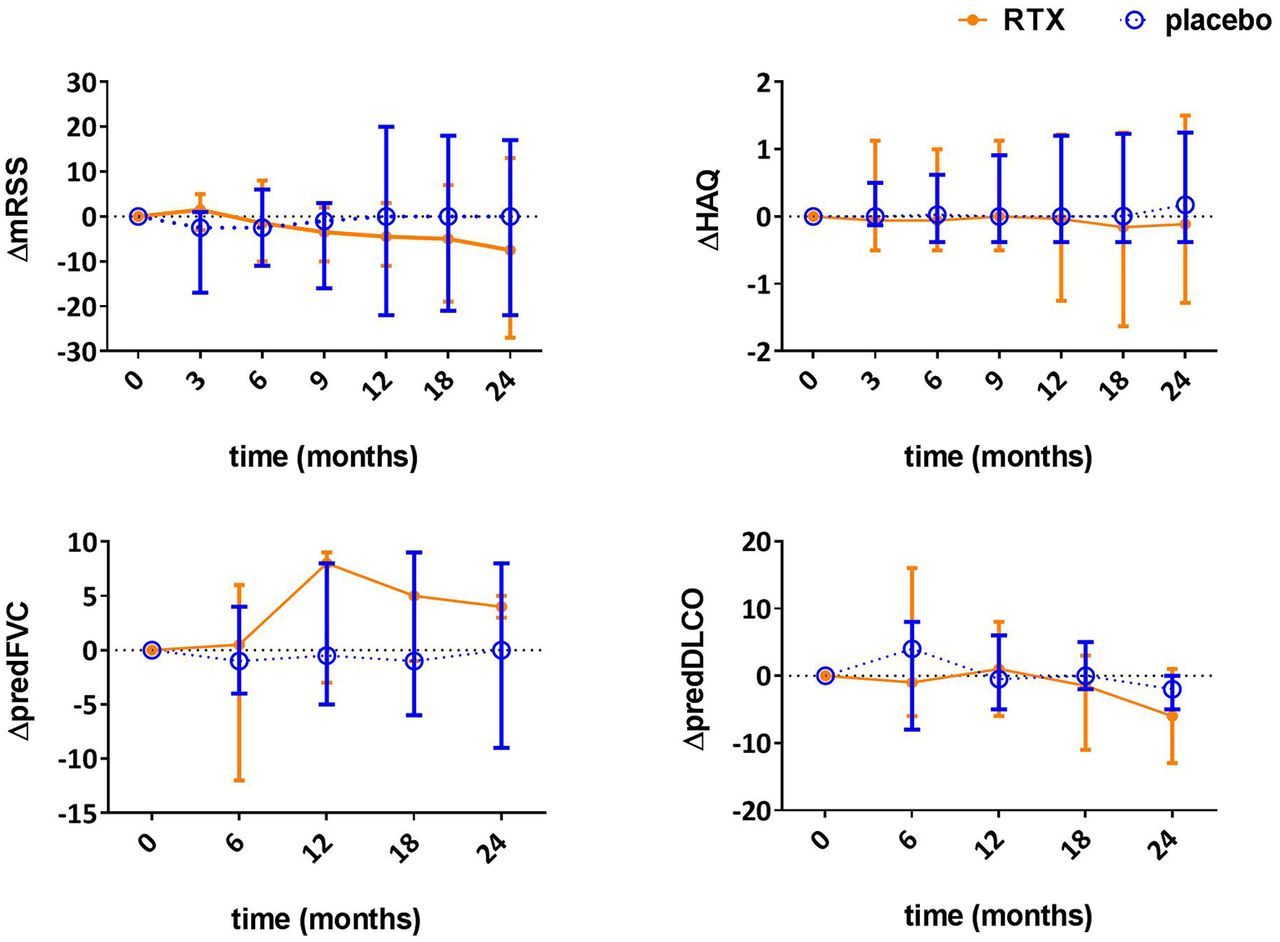
Change in modified Rodnan skin sore, forced vital capacity, diffusing capacity of the lung and HealthAssessment Questionnaire during 24-month follow-up in patients with systemic sclerosis treated with rituximab (RTX) orplacebo. HAQ, Health Assessment Questionnaire; RTX, rituximab; mRSS, modified Rodnan Skin Score; predFVC, forced vitalcapacity, percentage of predicted; predDLCO, diffusing capacity of the lung, percentage of predicted. Dots indicate medians;error bars indicate ranges.
Analysis of HRCT data according to Goh criteria showed a mean change in percentage of affected lung tissue between baseline and 12 months of −1.6% for RTX and +2.8% for placebo (p=0.28). Beneficial effects were explained by a decrease in ground glass opacities with RTX treatment in two RTX-treated patients (figures 3,4). Numerically, n=2/7 RTX patients and n=0/8 placebo patients showed improvement on HRCT (−10% or more change in mean extent of lung involvement), n=4/7 in RTX versus n=7/8 in placebo had stable lung involvement on HRCT (between −10% and +10% change in mean extent of lung involvement) and n=1/7 in RTX versus n=1/8 in placebo had worsening lung involvement on HRCT (+10% or more change in extent of lung involvement).
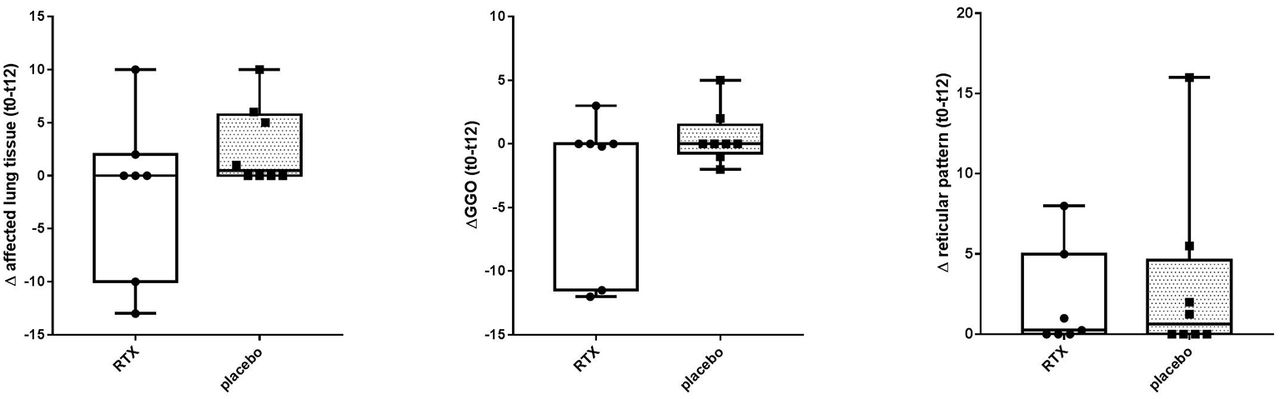
Change in mean extent of lung tissue and lung tissue with ground glass or reticular pattern involvement from baseline to 12 months follow-up in patients with systemic sclerosis. Vertical axis represent differences in Goh scores between baseline and 12 months for (from left to right): (1) mean extent of affected lung tissue, (2) GGO; mean extend of ground glass opacities, (3) mean extend of affected lung tissue with reticular pattern. At 12 months a non-significant trend in favour of rituximab was observed (mean change in lung involvement: -1.6% RTX group vs. +2.8% placebo group [p=0.28]). The beneficial effect in the rituximab group was explained by a decrease of ground glass opacities. RTX, rituximab; t0, baseline; t12, 12 months follow-up.

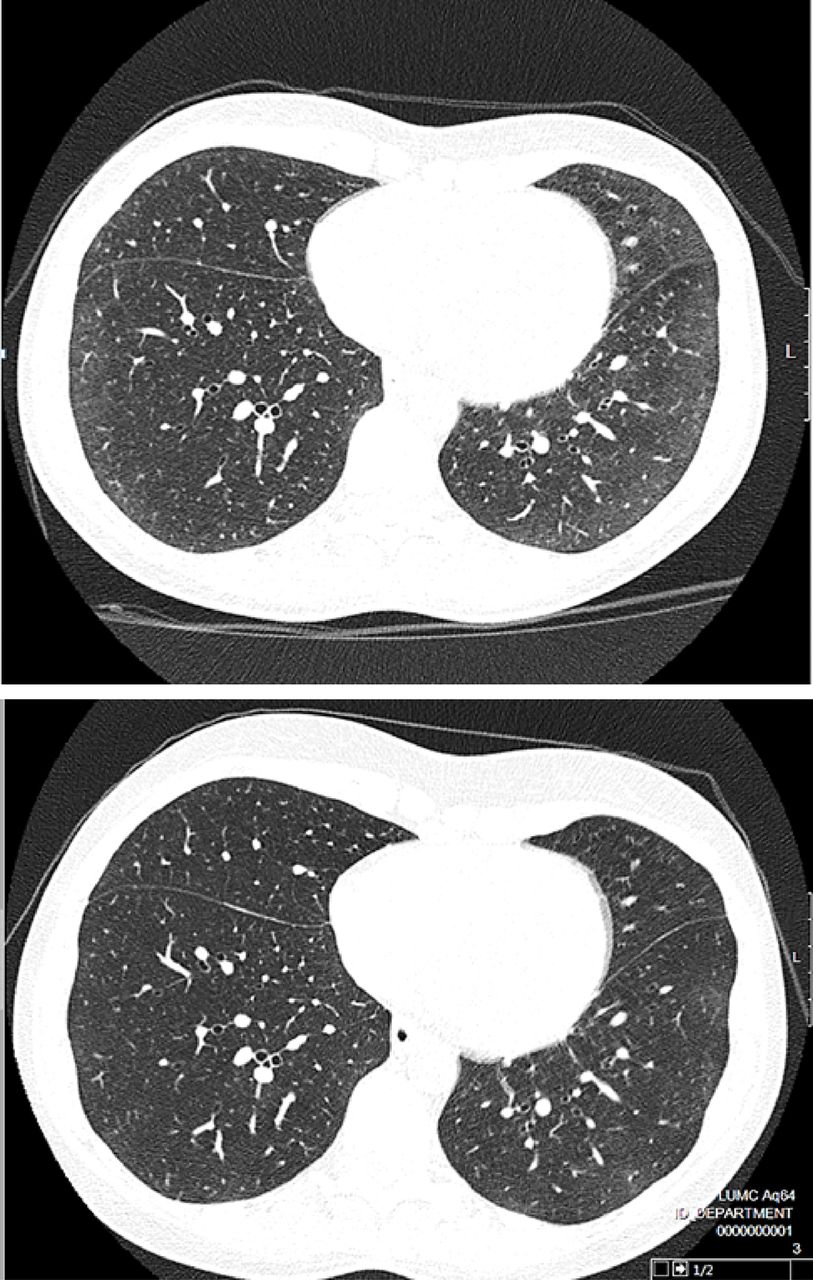
Improvement of lung involvement as evaluated by HRCT of a patient treated with rituximab. HRCT, high resolution computed tomography; inspiratory scan, halfway between pulmonary venous confluence and right hemi-diaphrag. Upper HRCT: baseline HRCT, before rituximab use; lower HRCT: 12 months after initial gift of rituximab.
Analysis of change in AUC showed no significant differences in the mRSS, FVC and HAQ-DI between the groups (see online supplementary table S2). Within the first year, mean change from baseline to 12 months follow-up in mRSS was comparable between groups, with −1.4 for the RTX and −2.7 for the control group (difference 1.3; 95% CI −3.4 to 6.2; p=0.55). For FVC and HAQ-DI, differences in AUC between baseline and 1 year were also insignificant. For FVC, there was a slight improvement with RTX and a slight deterioration with placebo (mean change AUC baseline to 12 months follow-up 0.6 for RTX and −0.4 for placebo, p=0.59). Also during the second year, no significant differences were observed in AUC for mRSS, FVC and HAQ-DI (see online supplementary table 2).
Efficacy analyses for the individual disease parameters were repeated excluding the two patients with disease duration of >24 months since diagnosis. These analyses did not show different results.
Recorded parameters reflecting vascular complications did not differ between the treatment arms: no patients in the trial had impaired kidney function and estimated glomelurar filtration rates (eGFR) within study arms were comparable (mean eGFR at T=0: placebo 118.5 mL/min/1.73 m2, RTX 106.8 mL/min/1.73 m2) and were stable throughout the trial. Also LVEF (as measured by echocardiography) remained stable throughout the trial in all participants (mean LVEF at T=0: placebo 62.0%, RTX 60.2%, at T=24: placebo 60.2%, RTX 65.5%). Digital ulcers occurred both in the placebo (n=3) and the rituximab group (n=3).
In the analyses of SF-36 scores and EQ-5D scores, no differences were seen (data not shown).
Two patients in the placebo group showed disease progression during follow-up according to prespecified criteria, including the patient that died after dropout. In the rituximab group, one patient showed disease progression. Apart from the patient that died, study disease progression was based on a ≥30% increased mRSS relative to baseline at the 12-month (placebo, n=1) and 18-month visits (RTX, n=1) in both cases. Including the patient that died after dropout, there was no difference in progression-free survival between groups (log rank (Mantel-Cox) p=0.674). Also after excluding the two patients with disease duration of >24 months, there was no significant difference in progression-free survival between the groups.
Safety and toxicity
No patients died during the study. One patient (placebo group) died due to disease progression, after dropout at 6 months because of active disease. This patient eventually died at 23 months due to scleroderma renal crisis after autologous haematopoietic stem cell transplantation.
A total of 88 adverse events (AEs) occurred during the study: 52 in the RTX group (six grade 3 AEs, two grade 4 AEs) and 36 in the control group (seven grade 3 AEs, zero grade 4 AEs) (p=0.22)(see online supplementary table S1). There were seven serious adverse events (SAEs) in the RTX group and four in the placebo group (p=0.36). SAEs in the RTX group were a breast carcinoma (18 months after first gift of RTX), abnormal cervical histology leading to hysterectomy (6 months after first gift of RTX; medical history of this patient mentioned abnormal cervical histology also before inclusion in the trial), an anaemia due to severe menstruation (7 months after first gift of RTX), a pancytopenia (12 months after first gift of RTX) and three events related to digital ulcers (n=2 at 1 month after first gift of RTX, the other at 7 months). SAEs in the placebo group included severe weight loss which required treatment by percutaneous endoscopic gastrostomy placement (17 months after first gift of RTX) and three events related to digital ulcers (1, 14 and 18 months after initial RTX). There were more grade 1 AEs in the rituximab group due to mild infusion reactions (system organ class type: immune system). A clear causal relation between AEs and treatment with RTX could not be established, except for mild infusion reactions.
Immunophenotyping of peripheral blood mononuclear cells
At baseline, there were no differences seen in proportions of macrophages (CD14+), NK cells (CD56+), T helper cells (CD3+/CD4+), cytotoxic T cells (CD3+/CD8+) or B cells (CD19+) between placebo and rituximab group (Data not shown). Three months after initial anti-CD20 treatment, significant depletion B cells was seen and simultaneously a decline in T cells was observed. Counterwise the proportion of macrophages increased.
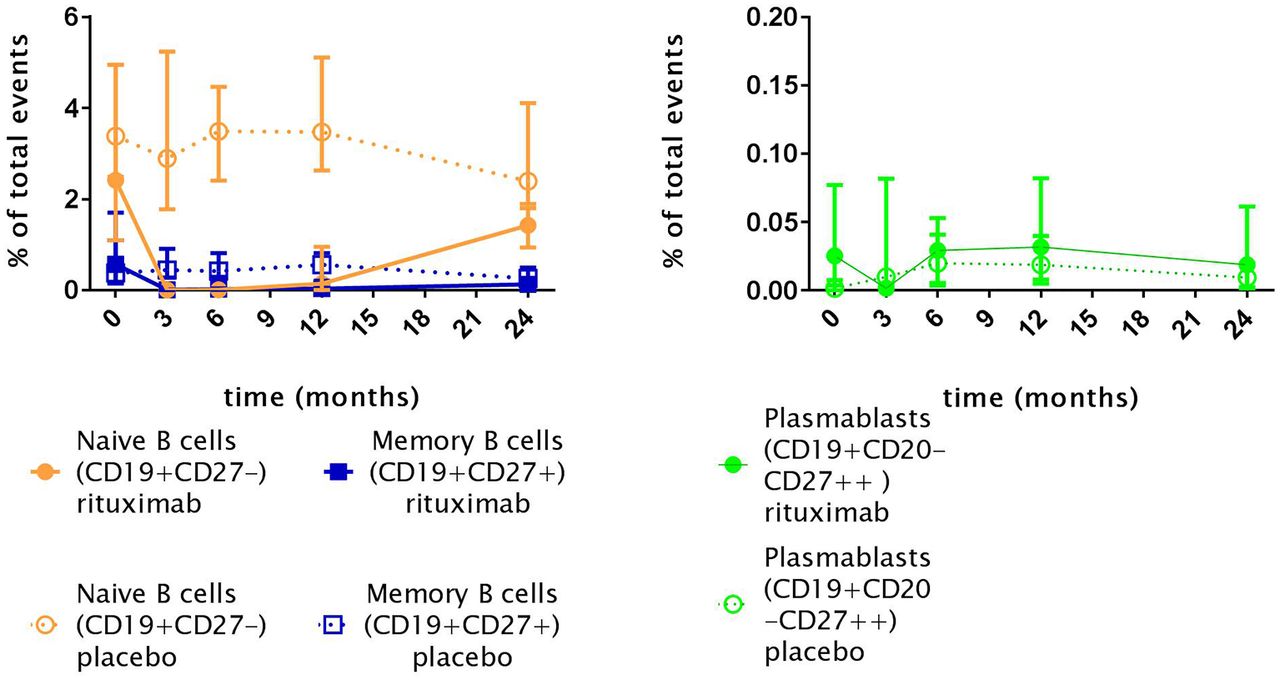
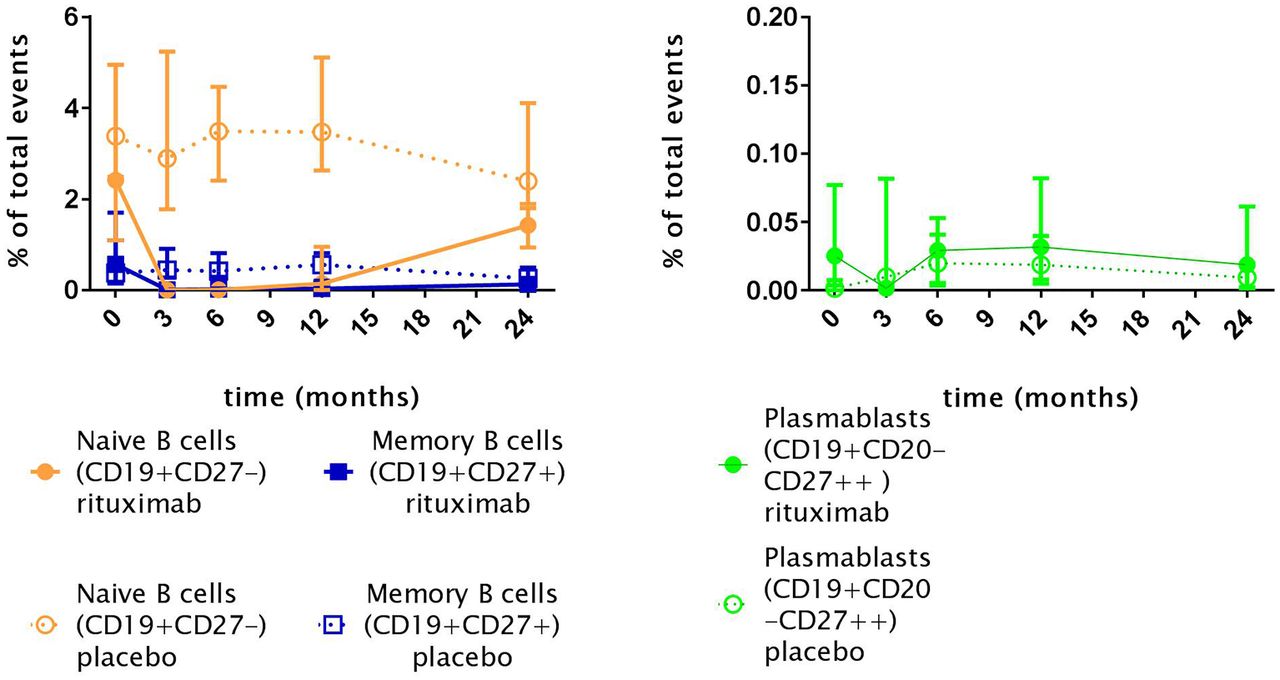
When observing the different subsets of B cells during the study, as shown in figure 5, naïve and memory B cells were depleted 3 months after the first gift of RTX. In five of eight patients with rituximab, a reduction of CD19+CD20-CD27++ plasmablasts was seen. Reduction of plasmablasts was significant within the RTX group when compared with baseline, but insignificant when compared with the placebo at the same time point. When assessing depletion of immunoglobulin expressing (IgG, IgA, IgM and IgD) naïve (CD19+CD27-) and memory (CD19+CD27+) subsets, as a positive control, all subsets were depleted (data not shown). At time of consolidation treatment (month 6), repopulation of naïve B cells, memory B cells and plasmablasts was present. However, throughout the complete follow-up period, repopulation of naïve B cells and memory B cells was incomplete in the RTX group (figure 5).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Naïve B cells (CD19+CD27-), memory B cells (CD19+CD27+) and plasmablasts (PB CD27++) levels in patients with systemic sclerosis. Indicated points resemble medians; Rituximab: T0, n= 8; T3 n=8; T6 n=7; T12 n=6; T24 n=4; Placebo: T0, n=8; T3, n=8; T6 n=8; T12 n=7; T24 n=7. Total events are defined as the number of detected cells by the flow cytometer. B cell depletion after rituximab treatment is seen in all B cell subsets.
ELISPOT data are presented in online supplementary figure 1, which shows persistence of plasma cells at month 3.
![[RITIS_-_2nd_REVISION_19-05-2017_Supp_Fig_1_-_ELISPOT.jpg]](https://rmdopen.bmj.com/content/rmdopen/3/2/e000384/DC2/embed/inline-supplementary-material-2.jpg?download=true){kind=link}
Skin biopsies
Skin biopsies were performed in 15 patients (RTX n=7, placebo n=8) at baseline and in 14 patients (RTX n=7, placebo n=7) at 3 months (see online supplementary table S3). For three patients in the placebo group, and for one patient in the RTX group, skin was clinically unaffected at the site of biopsy. Interobserver agreement of histological skin score evaluated by Fleiss’ kappa was κ=0.49 for T cells, κ=0.32 for B cells and κ=0.63 for macrophages.
There were no significant differences found in immune cell presence in skin neither between groups, nor within groups over time. At baseline there was a trend towards more mononuclear infiltration in the placebo group, based on the presence of more T cells. Over time, presence of T cells in the rituximab group increased at 3 months compared with baseline. Presence of other immune cells was stable over time (see online supplementary table S3).
B cells were rarely present in skin tissue, only one patient in the placebo group that showed a collection of >10 cells, but less than 50 cells at baseline. Scattered B cells (range 2–7 per biopsy) were seen in 5 out of 15 biopsies at baseline (RTX n=3, placebo n=2). Over time, there were no changes in the presence of B cells in skin of RTX-treated patients, 3 months after initial gift, with B cells present in 4 out of 7 biopsies. This was identical to the number of placebo patients with B cell presence in skin at 3 months (4 out of 7).
Discussion
This small randomised, placebo-controlled trial cannot reject nor confirm the hypothesis of RTX preventing fibrotic complications. No major safety issues were observed with RTX in the subset of patients with early SSc. Immunologically, RTX achieved its presumed biological effect: a depletion of circulating B cells up to minimal counts, but with persistence of antigen-secreting cells and incomplete depletion of the CD27++ plasmablast compartment. No change in the small number of cells from the B cell lineage present in skin tissue was observed with RTX treatment. Over time, small, non-significant differences in FVC, extent of pulmonary involvement and HAQ-DI in favour of the RTX group were found. Further research must confirm the credibility of these findings. A larger scale RCT in patients with proven pulmonary involvement therefore seems plausible and feasible.
Unfortunately, the trial had recruitment problems resulting in premature termination of inclusion. Moreover, patients in the control group experienced an unexpected favourable disease course, which complicates firm conclusions about efficacy of RTX in preventing fibrotic complications. This study aimed to include patients with early dcSSc. Indeed, our placebo group included patients of which the majority had dcSSc at baseline (63%), and four of eight patients were either ATA or RNApIII positive. Both these antibodies are associated with more severe disease course.43 44 Despite these characteristics reflecting high risk, early dcSSc, 75% of patients in the placebo group had favourable outcome after 2 years.
There is a small insignificant difference in disease duration between the RTX and the placebo group, with the placebo group having a longer disease duration. It has been shown that with longer disease duration, chances to improve spontaneously slightly increase45 which might partially explain the beneficial disease course in placebo. However, excluding the two patients with the longest disease duration did not change our results.
Several case reports, open-label studies and a nested case–control study thus far reported a potential beneficial effect of RTX on pulmonary function, skin fibrosis and functional impairment in SSc.12–21 46–49 Our observations are in line with the study from Lafyatis,15 who treated 15 patients with early SSc with RTX and did not find a clear beneficial effect on skin fibrosis and pulmonary function at 6 and 12 months of follow-up. Various explanations can account for the difference between previous open-label studies and our findings. As these studies did not include a placebo group, part of the observed efficacy might reflect natural disease course. In addition, most open-label studies included patients with longer disease duration13 14 50 and thus possibly selected an immunologically different subgroup of SSc patients. In comparison to the open-label studies of Lafyatis et al and Smith et al,15 17 who both also included patients with early disease, mean skin scores were lower in our population, which complicates the possibility of demonstrating clear clinical efficacy on skin involvement. When analysing only the patients with dcSSc at baseline, RTX n=3/7 showed a decrease in mRSS >5 versus n=2/5 within the placebo group. On the other hand, it is known that patients with more skin fibrosis at baseline are more likely to regress even without therapy over the next year.51
Immunophenotyping of peripheral blood showed almost complete B cell depletion, which is in line with previous studies.15 17 It is known that during treatment with RTX, plasmablasts and plasma cells can persist.52 Besides confirming this with ELISPOT, thereby showing persistence of IgA, IgG and IgM antigen-secreting cells after treatment with RTX, this is also demonstrated by the incomplete depletion of the CD27++ plasmablast compartment (see online supplementary figure S1). Other studies found CD20-positive B cells in skin biopsies in approximately half of patients at baseline and depletion in most of these cases.12 13 17 To overcome possible interference of RTX treatment with detection of CD20-positive B cells in skin, we chose to use CD79a staining on skin, which also stains plasmablasts and plasma cells that lack CD20, while in previous studies, CD20 staining was used. Based on morphology, the persisting B cells in our samples could reflect unaffected long-lived B cell populations. The exact nature and the relevance of these persistent B cells for development and persistence of skin fibrosis remains to be determined, and might be relevant in determining subsets of patients with SSc with high likelihood of responding to RTX.
Remarkably, two out of eight RTX-treated patients showed evident improvement of the extent of ground glass opacities in HRCT at 1 year follow-up versus none of the placebo-treated patients. Radiological improvement of CTD-associated and RA-ILD after treatment with RTX has also been described by other authors.53 54 Out of interest, and to possibly guide future research in the field, we compared different B cell subsets in baseline PBMCs between RTX patients with pulmonary improvement and those without. RTX-treated patients with pulmonary improvement both had higher counts of naïve B cells (CD19+,CD27-) (mean CD19+CD27+ of n=2 non-improvers under RTX 1.8% of total events vs n=6 improvers under RTX 5.6% of total events, p=0.003), while other subsets were comparable. We speculate that this subgroup of patients, possibly reflecting those with very early and active inflammatory pulmonary involvement, might be the subset of patients most likely to benefit from B cell depleting therapy. However, these observations obviously await replication.
In conclusion, we performed a double-blind placebo-controlled trial in patients with early SSc and show in-depth analysis of B cells in peripheral blood and skin tissue. Although given the small sample size and the unexpected favourable disease course in the placebo group, no firm conclusions on clinical efficacy of rituximab in early SSc can be drawn; our data show that a larger RCT in early SSc with proven pulmonary involvement might be worthwhile. In addition, inclusion of peripheral blood and skin tissue analyses is also warranted in future trials to determine the nature, role and relevance of persisting B cells in skin, and persisting plasmablasts and plasma cells in peripheral blood. Analysis as presented herein might help to identify a subset of patients with SSc most likely to benefit from B cell-depleting therapies.
Acknowledgments
We would like to thank Lucia Kroft, Department of Radiology LUMC for scoring HRCTs, the Department of Anatomy and Embryology LUMC for use of their aSMA antibody, Masson Trichrome staining (supplies) and microwave for immunohistochemistry staining and the Department of Pathology for their CD79a antibody.
References
Footnotes
Contributors TWJH, HUS, JKdVB, AS, NAM, MKN, JM all participated in study design and data collection. ALD, KDQ and MB performed immunohistologic scoring. AS and LK assessed HRCTs according to Goh criteria. NAM assessed cardiac echos. MB conducted statistical analyses. JKdVB and MKN helped with clinical interpretation. All authors contributed to refinement of the study protocol.
Funding This work was supported by Roche B.V., donating medication. Roche was not involved in study design; collection, analysis and interpretation of data; writing of the manuscript or in the decision to submit the manuscriptfor publication.
Competing interests MB is supported by a grant of Actelion Pharmaceuticals Nederland BV (Woerden, The Netherlands). ActelionPharmaceuticals had no role in the study design; collection, analysis and interpretation of data; writing of themanuscript or in the decision to submit the manuscript for publication.
Patient consent Detail has been removed from this case description/these case descriptions to ensure anonymity. The editors and reviewers have seen the detailed information available and are satisfied that the information backs up the case the authors are making.
Ethics approval Medical Ethical Committee (METC) of the Leiden University Medical Center.
Provenance and peer review Not commissioned; externally peer reviewed.