Article Text

Abstract
At the population level, rheumatoid arthritis (RA) is generally viewed as autoimmune in nature with a small subgroup of cases having a palindromic form or systemic autoinflammatory disorder (SAID) phenotype. Herein, we describe resistant cases of classical autoantibody associated RA that had clinical, genetic and therapeutic responses indicative of coexistent autoinflammatory disease. Five patients with clinically overlapping features between RA and SAID including polysynovitis and autoantibody/shared epitope positivity, and who had abrupt severe self-limiting attacks including fevers and serositis, are described. Mutations or single nucleotide polymorphisms in recognised autoinflammatory pathways were evident. Generally, these cases responded poorly to conventional Disease-modifying anti-rheumatic drugs (DMARD) treatment with some excellent responses to colchicine or interleukin 1 pathway blockade. A subgroup of RA cases have a mixed autoimmune-autoinflammatory phenotype and genotype with therapeutic implications.
- rheumatoid arthritis
- fever syndromes
- cytokines
- inflammation
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
A subgroup of rheumatoid arthritis (RA) cases can have a mixed autoimmune-autoinflammatory overlapping clinical phenotype which has implications for choice of appropriate therapy.
This points to innate immunity or autoinflammatory mechanisms as underpinning flare or relapse in some autoantibody positive RA cases.
These findings point to the marked genetic heterogeneity in RA cases that is thus far poorly recognised.
Introduction
Rheumatoid arthritis (RA) is genetically, phenotypically and molecularly heterogeneous. Most cases have an autoimmune phenotype as indicated by the major histocompatibility complex Class 2 (MHC-II) association, shared autoantibodies and responses to B cell depletion and co-stimulatory blockade therapies. However, there is growing evidence that innate immunity plays an important role in the pathogenesis of this disorder.1 We and others have also observed that some RA cases clinically resemble patients with so-called systemic autoinflammatory disorders (SAIDs),2 which would support this idea.
Broadly speaking SAIDs are diseases of the innate immunity caused by the mutations in genes including TNFRSF1A, NLRP3, MEFV and NOD2, which control the release of the proinflammatory cytokines, mainly interleukin (IL)-1ß, and IL-6 and Tumor necrosis factor (TNF).3 Typically, the patients experience abrupt systemic inflammatory attacks characterised by fever, prominent joint swelling, erythema and a raised C-Reactive protein (CRP). Furthermore, patients with SAID respond to therapies such as colchicine and anti-IL-1 biologics, which target the innate immune system. We present a series of five patients who have overlapping clinical features of RA and SAID.
Materials and methods
For detailed information regarding patients’ clinical assessment, and methods used for genetic and cytokine studies, please see online supplementary file 1.
Case 1
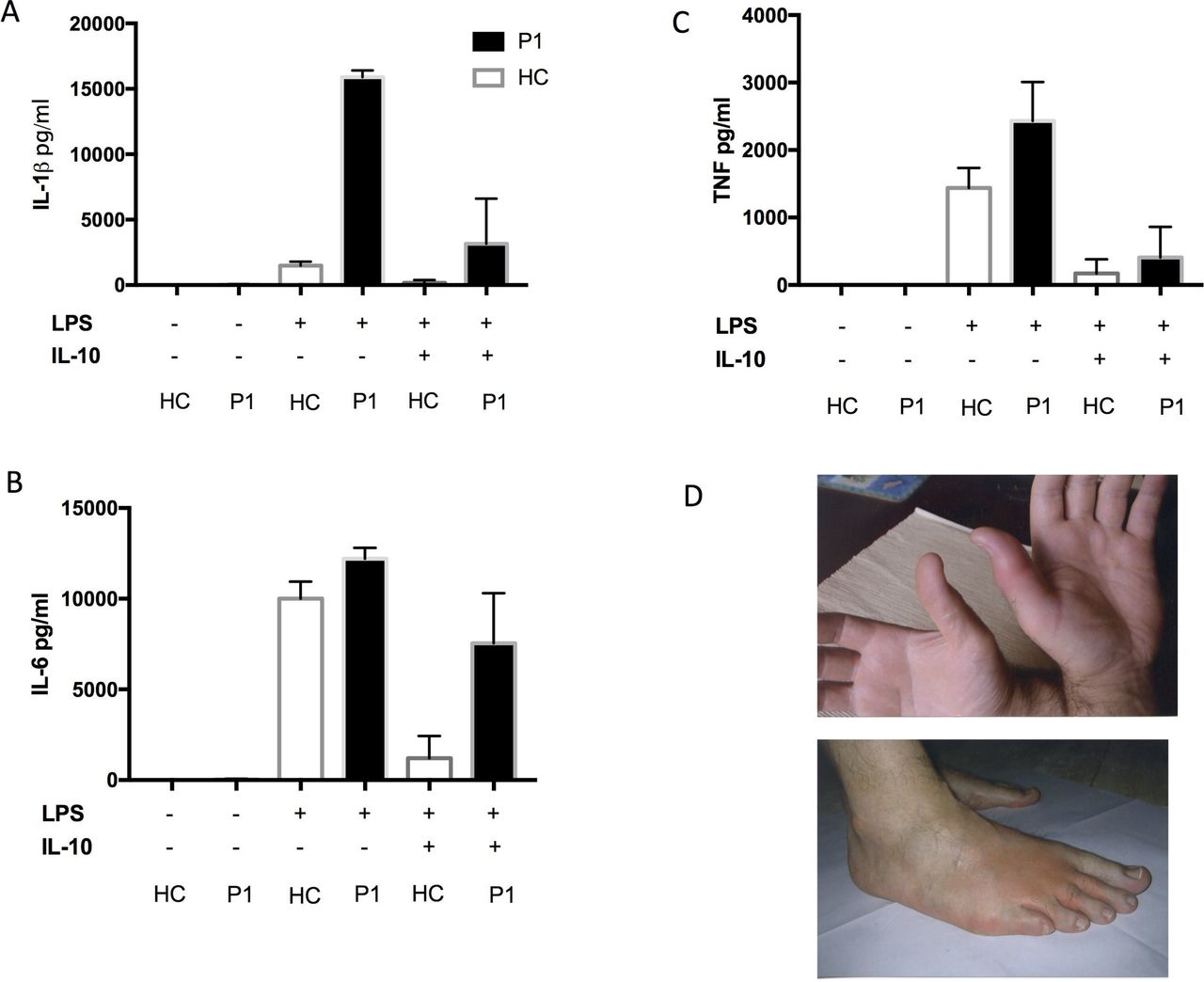
A 54-year-old man presented at the age of 35 years with episodic night sweats, fever, erythematous skin rash and bilateral symmetrical synovitis suggestive of RA (figure 1). Investigations showed elevated CRP and negative rheumatoid factor (RF), and skin biopsy had features in keeping with urticarial vasculitis. Over the next decade, he continued to suffer with episodic skin rashes and synovitis associated with systemic inflammation. He failed numerous empirical treatments including combinations of methotrexate and etanercept and methotrexate and tocilizumab (table 1). At the age of 47 years he was found to have a known pathogenic missense variant in the NLRP3 gene (p.Arg325Trp) consistent with diagnosis of cryopyrin associated periodic syndrome (CAPS).4 His lipopolysaccharide (LPS)-stimulated Peripheral mononuclear cells (PBMC) produced considerably higher levels of IL-1β compared with healthy controls. (figure 1). Anakinra was initiated (IL-1RA) in combination with methotrexate resulting in complete disease remission and normalisation of his CRP. As his methotrexate was withdrawn he had a major relapse of synovitis and developed clinical features of RA in association with anticitrullinated peptide antibodies (ACPAs) and RF positivity. His methotrexate was re-started and he again achieved clinical remission, but remains ACPA-positive.

Proinflammatory cytokine production from whole blood assay (case 1) in response to lipopolysaccharide (LPS) and after LPS/IL-10 combination (A,B,C). Picture of inflamed skin/joints, case 1 (D).
Summary of clinical features, investigations and treatments
Case 2
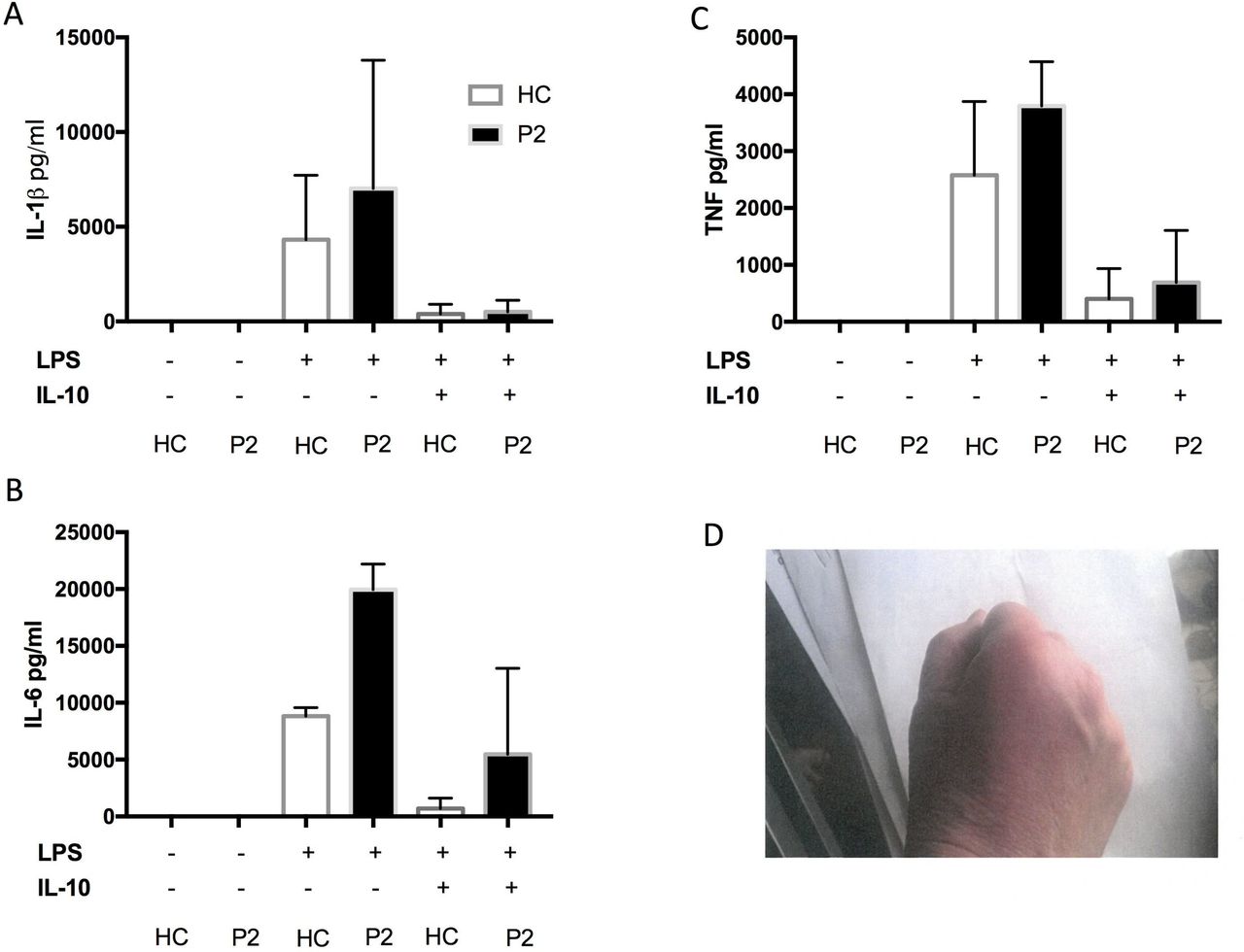
A 55-year-old woman presented at the age of 53 years with a 6 -month history of flitting palindromic arthropathy and raised inflammatory markers. Her symptoms would come on very abruptly and were accompanied by overlying soft tissue swelling and skin erythema. She also had a hospital admission for possible aseptic meningitis and later developed a blistering rash (negative antibullous Ab) and bilateral scleritis. She was found to be RF-positive and ACPA-positive. Genetic test revealed a compound heterozygote mutation in the MEFV gene c.289 C>T (Q97*) and c.605G>A (R202Q). Her PBMCs showed increased levels of IL-6 production following LPS stimulation, compared with healthy control. This response was poorly downregulated by IL-10 (figure 2). The patient had a modest benefit from colchicine but was unable to tolerate more than 500 µg/day. She responded well to a course of prednisolone and methotrexate, but remains dependent on low-dose prednisolone (7.5 mg).
{kind=link}
{kind=link}
Proinflammatory cytokine production from whole blood assay (case 2) in response to lipopolysaccharide (LPS) and after LPS/IL-10 combination (A,B,C). Picture of inflamed skin/joints, case2 (D).
Case 3
A 50-year-old man presented at the age of 49 years with a 12 -month history of synovitis, pleurisy, fever and pericarditis. He was initially diagnosed with polyarticular gout since the attacks of swelling around his foot were abrupt and associated with severe erythema. However subsequent investigations (table 1) were negative for gout and he was found to have high-titre ACPA and RF and a variant in NOD2 (c.2722G>C, p.Gly908Arg). He was originally managed with subcutaneous methotrexate but this was discontinued due to infective complications. Despite a reasonable initial response to colchicine and prednisolone, sulfasalazine was added to this combination as he continued to suffer from intermittent episodes of joint swelling associated with persistently raised CRP. The addition of sulfasalazine did not result in complete resolution of the attacks.
Case 4
A 42-year-old man at the age of 40 years developed severe right hip pain, with elevated CRP and neutrophilia. An aspirate was sterile and an MRI showed inflammation of the joint. He responded readily to diclofenac and antibiotic therapy, with complete resolution of his symptoms within 4 days. He had a similar presentation 5 months later in the other hip. The investigations showed both RF positivity and ACPA positivity and a polymorphism within the NOD2 gene c.2104 C>T (pArg702Trp). He benefited from an intramuscular corticosteroid pulse and was started on methotrexate, however the latter was discontinued because of tinnitus. Over the coming months, he developed clinical features of an asymmetrical polyarthritis which included episodes of marked knee swelling and an erythematous rash over affected joints. He was commenced on colchicine 500 µg twice daily resulting in complete resolution of his symptoms.
Case 5
A 60-year-old woman presented initially at the age of 43 years with metacarpophalangeal (MCP) joint swelling and a knee effusion. The synovial fluid analysis was negative. Over the following years she continued to suffer from fleeting episodes of severe MCP and knee joint swelling resulting in frequent attendance to acute medical services. There was never any documented fever or evidence of septic arthritis. Her clinical picture resembled RA with some atypical features. She was RF-positive but ACPA-negative and had elevated CRP during attacks. The genetic tests for SAID were negative. She received multiple treatments for presumed RA (table 1). However, the patient continued to experience acute, short-lived but disabling attacks of synovitis. She responded rapidly to colchicine with an almost instant cessation of her symptoms and stopped Disease-modifying anti-rheumatic drugs (DMARD) triple therapy.
Discussion
Herein, we report five cases of seropositive inflammatory arthritis, who met the European League Against Rheumatism/American College of Rheumatology diagnostic criteria for RA, but developed unusual clinical phenotypes resembling SAID. They all had abrupt onset of synovitis with associated joint erythema, excellent response to colchicine or anakinra, but poor response to traditional DMARDs. Genetic investigations showed that patients carried the DRB1 shared epitope, confirming their risk for developing RA and ACPA, and rare genetic variants associated with SAID.
There is only one previous report of concurrent CAPS and RA with a confirmed NLRP3 mutation.5 In our case we have also demonstrated enhanced IL-1ß release in response to LPS stimulation, which is typically associated with pathogenic gain-of-function NLRP3 mutations. The NLRP3 inflammasome, which is critical for release of active IL-1ß, has been linked to the pathogenesis of RA. The synovial expression of NLRP3 has positive correlation with the clinical and radiological arthritis scores in the collagen-induced model of arthritis.6 In human studies the genetic variation in NLRP3 inflammasome components as well as NLRP3 inflammasome activity have been shown to influence the susceptibility and severity of RA, and response to therapy.7–9
In case 2, two sequence variants in MEFV (pyrin) were found. R202Q is a frequent polymorphism, however Q97* is probably pathogenic according to the Infevers databse10 and associated with familial Mediterranean fever (FMF), a prototypical SAID known to respond to colchicine. A role for pyrin in pathogenesis of RA has recently been implied by a study showing that 14-3-3 protein, which is important in controlling the pyrin inflammasome activation,11 is detectable in sera of patients who later went onto develop seropositive RA.12 Therefore, two biological pathways that are intimately involved with processing of IL-1, a proinflammatory cytokine that defines the SAID field, also appear to have a role in the pathogenesis of RA. The IL-1β itself secreted by monocytes and macrophages have been shown to induce and amplify the inflammatory processes in the affected joints of patients with RA.13 Although anti-IL-1 blockade in RA has been inferior to anti-TNF/anti-IL-6, there is a subgroup of patients with RA with favourable response to anakinra. A study identified that higher disease severity, no or low dose steroid use, and concomitant diabetes were predictive of a response to anakinra.2 The authors therefore speculated that there might be two distinct groups of patients with RA, one with a more autoimmune phenotype associated with response to steroids and another with autoinflammatory characteristics defined by poor response to steroids but positive response to anakinra.
Cases 3 and 4 had variants in NOD2. Mutations in this gene are classically associated with Crohn’s disease; however neither patient had any evidence of inflammatory bowel disease. There is some evidence that this gene is activated in RA. For example, NOD2 was shown to be expressed in fibroblasts and synovium of patients with RA particularly at sites of invasion into the articular cartilage.14 Similarly, higher levels of NOD2 mRNA expression in PBMCs has been observed in patients with RA when compared with patients with osteoarthritis.15 However, cases 3 and 4 both had the alleles coding for a frequent polymorphism generally believed to be loss-of-function resulting in reduced nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) activation and lower inflammatory burden.16 The diagnosis of SAID in these cases was made on clinical grounds alone. Explanation for the clinical picture in these patients is therefore unclear, perhaps because of to tissue-specific genotype-phenotype associations.
Although genotyping was normal in case 5, clinically she had features of SAID and a most impressive response to colchicine. Recently myeloid restricted somatic mutations in NLRP3 have been shown to cause some acquired cases of SAID.17 These mutations cannot be detected using the standard genotyping methods, which may explain why in this case none were detected. In summary, this case series highlights that certain patients with RA may have a strong autoinflammatory component driving their disease process. Such patients may have disease which is refractory to conventional RA treatment and thus require a more tailored treatment strategy. This personalised approach to the spectrum of patients with RA is an area where further research is required.
Acknowledgments
The investigators thank their patients for giving consent.
References
Footnotes
Contributors All authors contributed towards writing of the study. SS, DM, PE and AGW contributed the cases. RD and GB-M conducted cytokine experiments.
Funding This work was supported by a grant form Sobi Pharmaceuticals, National Institute of Health Research (NIHR) Leeds Biomedical Research Centre, Cambridge Biomedical Research Centre and by grants from UNAM-DGAPAPAPIIT (IN217312 and IN220815) (GB-M).
Competing interests SS and DM have received honoraria from Sobi and Novartis for participation in advisory board meetings and for participating in pharma sponsored symposia.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement There are no additional unpublished data available from this study