Article Text

Statistics from Altmetric.com
Key messages
What is already known about this subject?
Transient receptor potential ankyrin 1 (TRPA1) is a membrane-associated cation channel primarily studied in sensory neurons, where it acts as a chemosensor of pungent compounds and mediates pain.
TRPA1 has recently emerged as a potential factor/mediator in arthritis; we have shown TRPA1 to mediate inflammatory and catabolic responses in primary human chondrocytes, as well as cartilage degradation, inflammation and pain in an experimental model of arthritis.
What does this study add?
This study shows that anti-inflammatory drugs dexamethasone and aurothiomalate downregulate the expression of functional TRPA1 in human chondrocytes.
How might this impact on clinical practice?
These results show a previously unknown mechanism of action for dexamethasone and aurothiomalate via downregulation of TRPA1, and thus provides a novel concept for the development of drugs for arthritis with analgesic and disease modifying properties.
Transient receptor potential ankyrin 1 (TRPA1) is a ligand-gated membrane-bound cation channel. TRPA1 has been largely studied in neurons, where it mediates pain and neurogenic inflammation and acts as a chemosensor for harmful exogenous compounds.1 2 More recently, TRPA1 has been found to be activated also by endogenous compounds formed in inflammatory conditions characteristic to arthritis, such as reactive oxygen and nitrogen species.3
We have recently discovered that TRPA1 is functionally expressed in primary human osteoarthritic chondrocytes,4 where it upregulated the production of mediators related to arthritis: interleukin (IL)-6, prostaglandin E2 and matrix metalloproteinases 1, 3 and 13.4 Furthermore, we showed in monosodium iodoacetate-induced experimental arthritis that TRPA1 activation mediates inflammation, cartilage degradation and pain.5 TRPA1 thus emerges as a novel factor associated with arthritis. Therefore, we investigated the effects of disease-modifying antirheumatic drugs methotrexate, sulfasalazine, hydroxychloroquine and aurothiomalate, glucocorticoid dexamethasone and non-steroidal anti-inflammatory drug ibuprofen on the expression of TRPA1 in human chondrocytes.
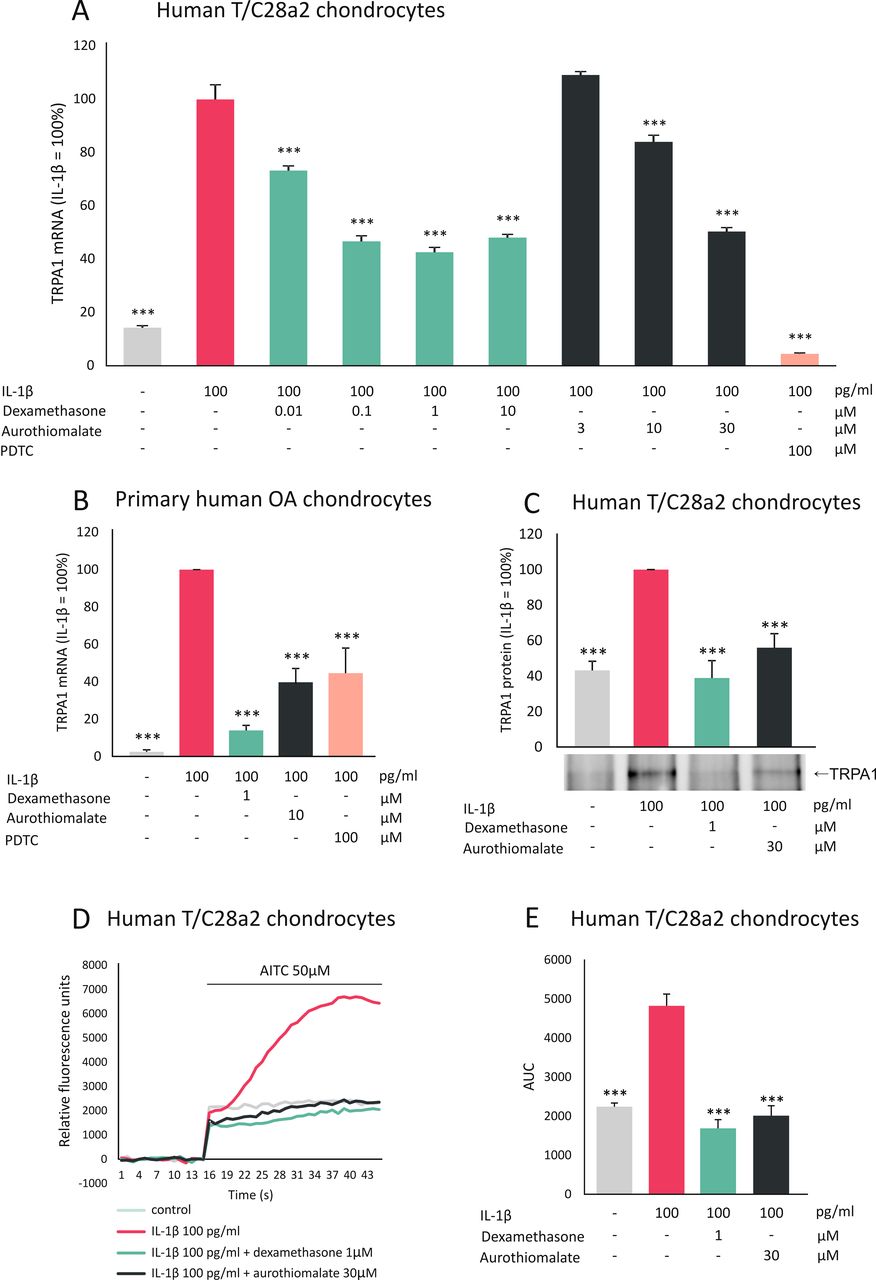
Human T/C28a2 chondrocytes6 were cultured with the aforementioned drugs, along with IL-1β, which was recently found to upregulate TRPA1 expression in chondrocytes.4 Dexamethasone and aurothiomalate inhibited TRPA1 mRNA expression in a dose-dependent manner (figure 1A), while methotrexate (10 µM), sulfasalazine (10 µM), hydroxychloroquine (10 µM) and ibuprofen (10 µM) had no effect. Dexamethasone and aurothiomalate also decreased TRPA1 protein levels (figure 1C).
{kind=link}
Dexamethasone and aurothiomalate inhibit TRPA1 expression in human chondrocytes. Human chondrocytes (primary human chondrocytes or T/C28a2 chondrocyte cell line) were cultured with IL-1β alone or in combination with the anti-inflammatory compound dexamethasone or aurothiomalate, or with the selective NF-κB inhibitor PDTC at concentrations given in the figure. (A, B) For TRPA1 mRNA expression analysis, human T/C28a2 chondrocytes (A) were incubated for 6 hours and the experiments were carried out in quadruplicate; primary chondrocytes (B) were incubated for 24 hours and the experiments were carried out in duplicate and repeated with cells from six donor patients. Total RNA was extracted and TRPA1 mRNA levels were measured by quantitative RT-PCR (TaqMan Gene Expression Assay for human TRPA1, Hs00175798_m1), and the results were normalised against GAPDH mRNA levels. (C) For TRPA1 protein analysis human T/C28a2 chondrocytes were incubated for 24 hours after which proteins were extracted and immunoprecipitated with TRPA1 antibody (SAB2105082, Sigma-Aldrich), and TRPA1 was detected with Western blot (with primary antibody: NB110-40763, NovusBiologicals). The figure shows one representative blot of six independent experiments with similar results. (D, E) For Ca2+ influx analysis human T/C28a2 chondrocytes were incubated with IL-1β alone or in combination with dexamethasone or aurothiomalate for 24 hours. Thereafter, the cells were loaded with Fluo-3-AM and the TRPA1-mediated Ca2+ influx was measured by Victor3 multilabel counter at excitation/emission wavelengths of 485/535 nm at 1/s frequency. In the measurements, basal fluorescence was first recorded for 15 s and thereafter the selective TRPA1 agonist AITC was added and the measurements were continued for 30 s. The results were normalised against the background and expressed as a mean of eight measurements. In (E), AUC from 15 to 45 s was calculated. Results are expressed as mean+SEM, results in (A–C) are expressed as a percentage in comparison to IL-1β-treated samples which were set as 100%. Statistical significance of the results was calculated with one-way or repeated measures ANOVA followed by Bonferroni post-test. Data were analysed using GraphPad InStat V.3.0. ***p<0.001. AITC, allyl isothiocyanate; ANOVA, analysis of variance; AUC, area under the curve; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IL, interleukin; NF-κB, nuclear factor kappa B; OA, osteoarthritic; PDTC, ammonium pyrrolidinedithiocarbamate; RT-PCR, reverse transcription PCR; TRPA1, transient receptor potential ankyrin 1.
Next, we examined if the observed drug effects on TRPA1 expression levels are functional, that is, if they are translated to changes in TRPA1-mediated calcium influx. In chondrocytes, which had been cultured in the presence of IL-1β, TRPA1-mediated calcium influx was increased; while that effect was reversed in cells which had been cultured with a combination of IL-1β and dexamethasone or aurothiomalate (figure 1D,E), confirming the functional downregulation of TRPA1 expression by these two drugs.
To confirm the effects of dexamethasone and aurothiomalate in primary human chondrocytes, cells were isolated from cartilage samples obtained from joint replacement surgery and cultured as described previously.4 Dexamethasone and aurothiomalate downregulated IL-1β-induced TRPA1 expression also in these primary chondrocytes (figure 1B).
The current results show for the first time that two anti-inflammatory drugs which are effective in the treatment of arthritis and retard cartilage degradation, namely dexamethasone and aurothiomalate, downregulate the expression of TRPA1 in human chondrocytes. Notably, inhibition of the transcription factor nuclear factor kappa B (NF-κB) also downregulated TRPA1 expression (figure 1A,B). Accordingly, Hatano et al7 reported recently that the TRPA1 promoter has at least six putative NF-κB binding sites; they also showed NF-κB to be involved in the induction of TRPA1 in synoviocytes. Glucocorticoids8 as well as aurothiomalate9 have been shown to inhibit NF-κB activation. Therefore, the downregulation of TRPA1 expression by these drugs may occur, at least in part, via inhibition of NF-κB.
The present findings, together with previous results,4 5 7 strongly suggest TRPA1 as a novel factor and drug target in arthritis.
Acknowledgments
We wish to thank Ms Terhi Salonen, Heini Brander and Ella Lehto for excellent technical assistance and Ms Heli Määttä for skilful secretarial help.
Acknowledgments
We wish to thank Ms Terhi Salonen, Heini Brander and Ella Lehto for excellent technical assistance and Ms Heli Määttä for skilful secretarial help.
Footnotes
Contributors EN, MH, LJM, TM, KV and EM contributed to the design of the study and to the acquisition, analysis and interpretation of data. EM conceived and supervised the study. EN drafted the manuscript. All authors revised the manuscript critically for important intellectual content and have approved its final version for submission.
Funding The study was supported by grants from the Competitive Research Funding of the Pirkanmaa Hospital District, Finland; Finnish Cultural Foundation, Finland; Research Foundation of Rheumatic Diseases, Finland; and Patient Organization for Rheumatoid Arthritis (Tampereen Reumayhdistys), Finland.
Competing interests None declared.
Patient consent Written informed consent was obtained from all patients.
Ethics approval Ethics Committee of the Pirkanmaa Hospital District, Finland.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All the data is reported in the manuscript.