Article Text

Abstract
Background Secukinumab treatment has previously been shown to significantly improve the signs and symptoms of active ankylosing spondylitis (AS), with responses sustained through 2 years. Here, we report the long-term (3 years) efficacy and safety of secukinumab in the MEASURE 2 study.
Methods MEASURE 2 (NCT01649375) is a 5-year phase III, randomised, double-blind, double-dummy, parallel-group, placebo-controlled study to evaluate the efficacy, safety and tolerability of subcutaneous loading and maintenance dosing of secukinumab in adult subjects with active AS. Subjects were randomised to receive subcutaneous secukinumab 150 mg, 75 mg or placebo at baseline, weeks 1, 2 and 3 and every 4 weeks from week 4. At week 16, placebo-treated subjects were rerandomised to receive secukinumab 150/75 mg.
Results Retention rates were high during weeks 16–156 and were 86% and 76% for secukinumab 150 and 75 mg, respectively. Secukinumab 150 mg provided sustained improvements in the Assessment of Spondyloarthritis International Society ASAS 20/40 response rates at week 156 (70.1%/60.9%) compared with week 52 (74.2%/57.0%); however, there was a slight decrease for secukinumab 75 mg (54.3%/37.0% vs 62.5%/43.2%, respectively). Sustained improvements were observed in all other end points, including Bath Ankylosing Spondylitis Disease Activity Index, AS Disease Activity Score with C reactive protein inactive disease, ASAS 5/6, Short Form-36 Physical Component Summary and ASAS partial remission. Clinical benefits were observed regardless of prior exposure to anti-tumour necrosis factor agents. The safety profile remained favourable and was consistent with previous reports.
Conclusions This study showed sustained improvement through 3 years in signs, symptoms and physical function in subjects with AS. Retention rates were high and secukinumab was well tolerated, with a favourable safety profile.
- ankylosing spondylitis
- cytokines
- dmards (biologic)
- spondyloarthritis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Secukinumab, a fully human monoclonal antibody to interleukin-17A, has shown efficacy in the treatment of inflammatory diseases such as psoriasis, psoriatic arthritis and ankylosing spondylitis (AS)
What does this study add?
This report presents the 3-year results of the MEASURE 2 study, which revealed that secukinumab provided sustained benefits in the signs and symptoms of ankylosing spondylitis, as well as improving physical function through 3 years in subjects with AS, with a retention rate >80%
Clinical benefits with secukinumab were observed regardless of prior exposure to anti-tumour necrosis factor (anti-TNF) therapy, with greater responses demonstrated in anti-TNF-naïve subjects
Secukinumab was well tolerated with a favourable safety profile, and no new safety concerns were identified.
How might this impact on clinical practice?
These data support the use of interleukin-17 inhibition with secukinumab for patients who fail conventional treatments, such as non-steroidal anti-inflammatory drugs or local glucocorticoids, and for patients who fail anti-TNF therapy
Introduction
Ankylosing spondylitis (AS) is a chronic inflammatory disease that can lead to progressive, irreversible structural damage of the spine, sacroiliac and/or peripheral joints; disability and reduced quality of life.1 Anti-tumour necrosis factor (anti-TNF) agents are effective in relieving the signs and symptoms of AS2–4; however, many patients experience an inadequate response or intolerance, relapse of disease on discontinuation, or safety concerns with anti-TNF therapy.5 6
Interleukin (IL)-17A, a pro-inflammatory cytokine produced by T helper 17 and other cells, is a key therapeutic target for the treatment of AS.7 Secukinumab, a fully human monoclonal antibody that selectively neutralises IL-17A,8 has been shown to have significant efficacy in the treatment of AS,8 9 moderate-to-severe psoriasis10 and psoriatic arthritis,11–13 demonstrating a rapid onset of action and sustained responses with a favourable safety profile.14 15 Secukinumab treatment has previously been shown to significantly improve the signs and symptoms of active AS in the phase III MEASURE 2 study (NCT01649375), with responses sustained through week 104.16 Here, we report the longer term (156 weeks) efficacy and safety of secukinumab treatment in MEASURE 2.
Methods
Study design
The MEASURE 2 study design, methodology and statistical analysis have been described previously.9 Briefly, this 5-year phase III trial uses a randomised, double-blind, double-dummy, parallel-group, placebo-controlled design to evaluate the efficacy, safety and tolerability of subcutaneous loading and maintenance dosing of secukinumab in subjects with active AS. Subjects with active AS were randomised to receive subcutaneous secukinumab 150 mg, 75 mg or placebo at baseline, weeks 1, 2 and 3 and every 4 weeks from week 4. At week 16, placebo-treated subjects were re-randomised to receive subcutaneous secukinumab 150 or 75 mg every 4 weeks, irrespective of the clinical response. Unblinding occurred after the week 52 analysis was performed, following which subjects continued to receive the same active dose of secukinumab as open-label treatment. After approval of a protocol amendment, any subject treated with secukinumab 75 mg by subcutaneous injection every 4 weeks could be up-titrated to 150 mg subcutaneous injection every 4 weeks if the investigator felt their overall therapeutic response was not fully achieved with the lower dose and that they might benefit with the higher dose.
Subjects
Subjects included in the study were aged ≥18 years and had active AS fulfilling the Modified New York Criteria,17 with a score of 4 or higher (scores range from 0 to 10, with higher scores indicating more severe disease activity) on the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI),18 and a spinal pain score ≥4 cm or more on a 10 cm Visual Analogue Scale (VAS) (with higher numbers indicating greater disease activity), despite treatment with the maximum tolerated doses of non-steroidal anti-inflammatory drugs (NSAIDs). Subjects previously treated with disease-modifying antirheumatic drugs and anti-TNF agents were included with prior washout periods. Subjects were included if they had an inadequate response to an approved dose of no more than one anti-TNF agent for 3 months or more, or had experienced unacceptable adverse effects. Concomitant sulfasalazine (≤3 g/day), methotrexate (≤25 mg/week), prednisone or equivalent (≤10 mg/day) and NSAIDs were permitted. Key exclusion criteria were total spinal ankylosis, evidence of infection or malignancy on chest X-ray, known HIV or hepatitis B or C infection at screening, active systemic infection within 2 weeks before baseline and previous treatment with cell-depleting therapies or biologic agents other than anti-TNF agents. The study was conducted in accordance with the Declaration of Helsinki. Written informed consent was provided by all enrolled subjects.
End points and assessments
Subjects initially randomised to secukinumab and those who switched from placebo to secukinumab at week 16 were combined for analyses after week 52 (secukinumab 150 mg, n=106 and secukinumab 75 mg, n=105). A total of 5 subjects up-titrated starting at week 140; these are included in the efficacy and safety analyses at week 156 in the treatment group they were originally randomised to. The primary end point was Assessment of Spondyloarthritis International Society criteria (ASAS) 20 at week 16. The ASAS 20 response was defined as an improvement of ≥20% and ≥1 unit (on a 10-unit scale) in at least three of the four main ASAS domains (subjects’ global assessments of disease activity, pain, physical function and inflammation) and no worsening of ≥20% and ≥1 unit (on a 10-unit scale) in the remaining domain. The ASAS 20 response was measured through week 156. Other assessments included: ASAS 40 response; AS Disease Activity Score with C reactive protein (ASDAS-CRP); ASAS 5/6; BASDAI; ≥50% improvement in the baseline total BASDAI score (BASDAI 50 response); Short Form-36 Physical Component Summary (SF-36 PCS) and ASAS partial remission. The overall safety and tolerability of secukinumab was reported through week 156. Routine safety monitoring was performed; treatment-emergent adverse events (AEs) and serious AEs are reported.
Statistical analyses
Data on efficacy are reported as observed through week 156 by study treatment dose. Summary statistics of the categorical variable are based on observed frequencies and percentages; for continuous variables, the mean ± SD or median (range) are used as appropriate. Analyses stratified by anti-TNF history (anti-TNF-naïve vs inadequate response or intolerance to an anti-TNF agent (anti-TNF-IR)) were prespecified and reported as observed. Following week 52, subjects initially randomised to secukinumab and those who switched from placebo to secukinumab at week 16 were combined. Subjects that up-titrated were counted only in their originally randomised treatment group. For patients who discontinued during the period from week 108 to 156 (ie, current treatment period for this analysis), the end of treatment visit (ie, final assessment 4 weeks after last study treatment) was considered as week 156. Safety analyses included all subjects who received ≥1 dose of the study treatment.
Results
Subjects
A total of 219 subjects with active AS were randomised at baseline to subcutaneous secukinumab 150 mg (n=72), 75 mg (n=73) or placebo (n=74) (20). At week 16, 66 placebo-treated subjects were rerandomised to subcutaneous secukinumab 150 mg or 75 mg (eight placebo-treated subjects discontinued the study before week 16). Between week 16 and 156, subject retention rates were 86% (86/100) and 76% (76/100) for secukinumab 150 and 75 mg, respectively. The higher discontinuation rate for 75 mg was in part due to lack of efficacy or subject/guardian decision (figure 1). Demographic and baseline characteristics were similar to the core trial population (table 1), which has been reported in detail previously.9

Subject disposition through week 156 of treatment. aIncludes placebo switchers, who were rerandomised at week 16; bIncludes patients who up-titrated from secukinumab 75 to 150 mg at week 140.
Demographics and baseline disease characteristics
Efficacy
Results of the primary end point, the proportion of subjects who met ASAS 20 response criteria at week 16, have been previously reported.9 At week 156, ASAS 20/40 response rates were 70.1%/60.9% and 54.3%/37.0% for secukinumab 150 and 75 mg, respectively (figure 2A and B; data shown as observed). These were sustained from week 52, where ASAS 20/40 response rates were 74.2%/57.0% and 62.5%/43.2% for secukinumab 150 and 75 mg, respectively.
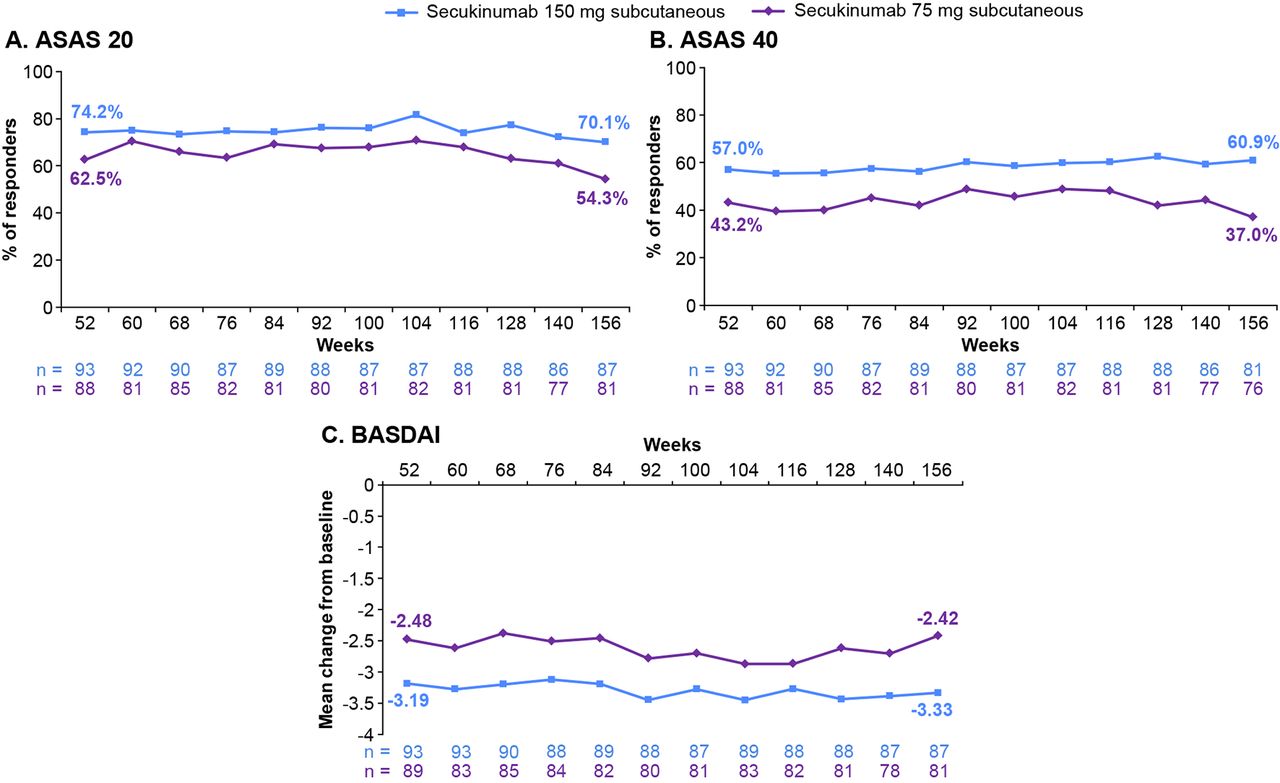
ASAS 20/40 response rates, and mean change from baseline in BASDAI through week 156* of treatment. *For patients who discontinued, the end of treatment visit (ie, final assessment 4 weeks after last study treatment) was considered as week 156. Data are shown as observed through week 156. ASAS 20/40, Assessment of Spondyloarthritis International Society criteria for 20%/40% improvement; BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; n, number of subjects in the treatment group with evaluation at each time point.
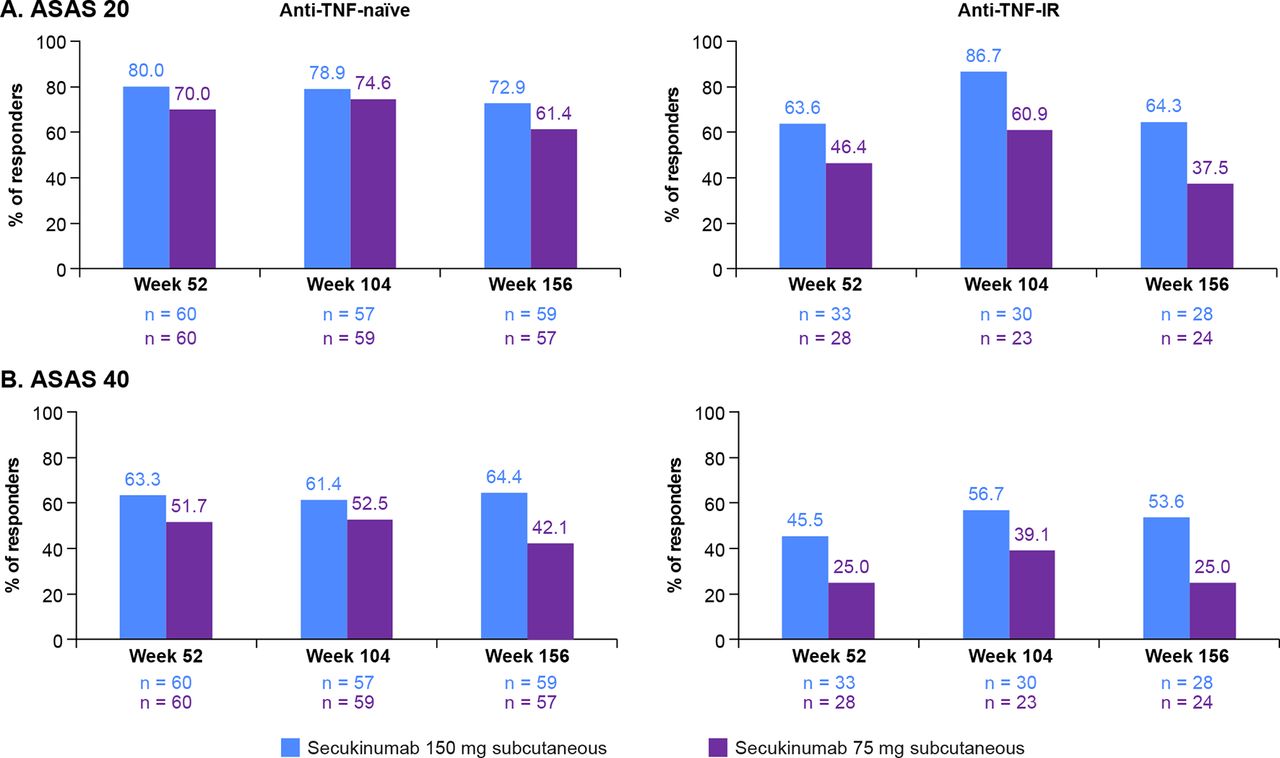
ASAS 20/40 response rates by prior anti-TNF status at weeks 52, 104 and 156 are shown in figure 3. At week 156, ASAS 20/40 response rates in anti-TNF-naïve subjects were 72.9%/64.4% and 61.4%/42.1% in the secukinumab 150 and 75 mg groups, respectively. In contrast, in anti-TNF-IR subjects, ASAS 20/40 response rates were 64.3%/53.6% and 37.5%/25.0% in the secukinumab 150 and 75 mg groups, respectively.
{kind=link}
{kind=link}
{kind=link}
ASAS 20/40 responses in anti-TNF-naïve and anti-TNF-IR subjects at weeks 52, 104 and 156. Data are shown as observed through week 156. ASAS 20/40, Assessment of Spondyloarthritis International Society criteria for 20%/40% improvement; IR, inadequate response; n, number of evaluable subjects; TNF, tumour necrosis factor.
The mean change from baseline in BASDAI was sustained from −3.2 at week 52 to −3.5 at week 104, and −3.3 at week 156 with secukinumab 150 mg (figure 2C); similar findings were observed for the 75 mg dose. Clinical improvements with secukinumab treatment were sustained through week 156 across other end points, including ASAS 5/6, SF-36 PCS and BASDAI50 response (table 2); these improvements were observed regardless of previous anti-TNF exposure. The proportion of subjects meeting the criteria for ASDAS-CRP inactive disease increased from 19.4% at week 52 to 24.1% at week 156 with secukinumab 150 mg, but decreased from 17.2% to 14.8% with secukinumab 75 mg. Similar observations were made for those achieving ASAS partial remission (table 2).
Clinical improvements with secukinumab overall, and in anti-TNF-naïve and anti-TNF-IR subjects at weeks 52 and 156
Safety
The total exposure to secukinumab over the entire treatment period was 914.3 patient-years. The safety and tolerability profile of secukinumab was consistent with previous reports; nasopharyngitis, upper respiratory tract infection and diarrhoea were the most frequently reported AEs. Exposure-adjusted incidence rates (per 100 patient-years) for AEs of special interest with any secukinumab dose were serious infections and infestations (1.5), Crohn’s disease (0.6), ulcerative colitis (0.6), undifferentiated inflammatory bowel disease (0.2), Candida infections (1.0) and major adverse cardiovascular events (0.6). No cases of tuberculosis reactivation, opportunistic infections or suicidality-related AEs were reported. Three deaths were reported: two in the secukinumab 75 mg dose (one acute myocardial infarction in a man aged 60 years before week 52; one case of respiratory arrest due to pneumonia in a man aged 77 years between week 104 and 156), and one in the secukinumab 150 mg dose (pneumonia, cardiac hypertrophy and dilation in a man aged 39 years between week 104 and 156); none was suspected to be related to study treatment.
Discussion
In the previously published results of the MEASURE 2 study, secukinumab 150 mg showed significant efficacy versus placebo in the primary end point, the proportion of subjects who met ASAS 20 response criteria at week 16 (61.1% vs 28.4%), as well as the other prespecified end points (ie, ASAS 40 response rates, BASDAI, ASDAS-CRP inactive disease, ASAS 5/6 and SF-36 PCS), except ASAS partial remission.9 Although the 75 mg dose did show increased efficacy versus placebo at week 16 (41.1% vs 28.4% for ASAS 20), it did not reach statistical significance based on the hierarchical hypothesis testing that controlled for multiplicity of testing.9 In the present analysis, clinical improvements were sustained through week 156 across all measured end points for secukinumab 150 mg, thus supporting the long-term suitability of secukinumab for controlling the signs and symptoms of AS.
Sustained improvements were observed in subjects who switched from placebo to secukinumab, and in anti-TNF-naïve and anti-TNF-IR groups through week 156. Indeed, the proportion of subjects meeting ASAS 40 response criteria and ASAS partial remission increased for secukinumab 150 mg from week 52 to week 156 in both anti-TNF-naïve and anti-TNF-IR groups, with higher increases for both measures in anti-TNF-IR subjects. The higher rates of increase in ASAS 40 and ASAS-PR in patients who were anti-TNF-IR compared with those who were anti-TNF-naïve could be a result of a number of factors, including the heterogeneity of the anti-TNF-IR subpopulation; this comprised patients who previously failed anti-TNF treatment for any one of several reasons, including lack of primary or secondary efficacy, intolerance or safety concerns. These improvements in subjects in whom anti-TNF therapy had previously failed suggests that secukinumab 150 mg could offer symptom relief for this population who are in need of an alternative therapy to anti-TNF agents.
This report confirms that secukinumab was well tolerated through 3 years of treatment, with no new safety signals or unexpected safety findings. Findings were consistent with previous studies of secukinumab for ankylosing spondylitis, including the independent MEASURE 1 trial, which reported that intravenous loading of secukinumab 150 or 75 mg provided sustained efficacy in signs, symptoms and physical function in subjects with AS over 3 years, with no new safety concerns identified.9 19 Similar sustained improvements have been seen in anti-TNF-treated patients; however, TNF-antagonists are not efficacious in some patients and have some safety concerns.5 20 21 AEs through week 156 were mostly mild to moderate in severity, and primarily driven by non-serious infections.
Limitations of the current analysis include the lack of a comparator group beyond week 16 and the fact that subjects and investigators were aware that all subjects received secukinumab from week 16 onwards, which could introduce systematic bias in the reporting of results. This study was also limited in that it was not designed to identify a difference between doses or to assess differences in response according to previous anti-TNF use.
Conclusions
Building on earlier findings, these results show sustained improvement through 3 years in signs, symptoms and physical function in subjects with AS, who remained on subcutaneous secukinumab 150 mg. Retention rates were high and secukinumab was well tolerated with a favourable safety profile, consistent with previous reports.
Acknowledgments
Medical writing and co-ordination support was provided by Martin Wallace and John Gallagher, of Novartis, in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
References
Footnotes
Contributors All authors were involved in the study design and/or collection, analysis and interpretation of the data, provided critical revision of the manuscript and approved the final version to be submitted for publication.
Funding This study was funded by Novartis Pharma AG, Basel, Switzerland.
Competing interests HM-O: grant/research support from Janssen and Celgene; speaker fees from Janssen, Pfizer, AbbVie, Celgene, Novartis and UCB. JS: grant/research support from AbbVie, Pfizer and Merck, consultant for AbbVie, Pfizer, Merck, UCB and Novartis; speaker support from: AbbVie, Pfizer, Merck and UCB. AK: grant/research support from Altoona Center for Clinical Research, PC; consultant fess from Vertex, AbbVie, Amgen, Celgene, Horizon, Genetech, Janssen, Merck, Novartis, Pfizer, UCB, Genzyme, Sanofi, Regeneron, SUN Pharma Advanced Research, Boehringer Ingelheim. RB: grants/research support from AbbVie, MSD and Roche; consultant fees/speaker support from AbbVie, Pfizer, Roche, Bristol-Myers, Janssen, Lilly and MSD. MC: consultant fees/speaker support from AbbVie, Amgen, BMS, Celgene, Janssen, Lilly, Merck, Novartis, Paladin, Pfizer, Roche, Sanofi, UCB. EMD: employee of Novartis. SR: employee of, and owns stock, in Novartis. HR: employee of, and owns stock, in Novartis.
Ethics approval The MEASURE 2 study protocol and all amendments were reviewed by the Independent Ethics Committee and Institutional Review Board at each center before the start of the study.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All relevant data are within the text.