Article Text

Abstract
In the late 1980s, the description by Modic and colleagues of elementary discovertebral changes detected on MRI (Modic classification) suggested for the first time a possible correlation between anatomical and clinical features in a subgroup of patients with non-specific chronic low back pain. Degenerative disc disease is frequent and usually asymptomatic, but Modic 1 changes in the vertebral endplates adjacent to a degenerated disc are associated with inflammatory-like chronic low back pain and low-grade local and systemic inflammation, which led to the concept of ‘active discopathy’. Active discopathy shares some similarities with acute flares of peripheral osteoarthritis. Likewise, what triggers disc activation and how it self-limits remain unknown. A better understanding of mechanisms underlying disc activation and its self-limitation is of clinical relevance because it may enable the design of more targeted pharmacological and non-pharmacological interventions for the subgroup of patients with chronic low back pain and active discopathy. Here, we narratively review current disc-centred biomechanical and biochemical hypotheses of disc activation and discuss evidence of interactions with adverse personal and environmental factors.
- low back pain
- rehabilitation
- magnetic resonance imaging
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Active discopathy is a specific entity that associates clinical, biological and MRI signs of activity with degenerative disc disease.
What does this study add?
The onset of active discopathy is unknown but is most likely multifactorial, involving both biomechanical and complex biochemical adverse factors in addition to genetic and individual predisposing factors.
How might this impact on clinical practice?
Although local inflammation is currently the main therapeutic target, it may be only a late-stage marker of disc disease activation, and a better understanding of disc activation complex pathogenesis is needed to develop more comprehensive and long-term efficient treatments.
Introduction
Non-specific low back pain (LBP) is the first cause of years lived with disability.1 The aetiological diagnosis of LBP is challenging because a consistent anatomoclinical correlation is usually lacking. However, the development of lumbar MRI and studies of large cohorts of patients with LBP have allowed for isolating lesions involving the vertebral endplates adjacent to degenerative disc disease (DDD) that seem closely related to persistent painful symptoms.2 3 These lesions were classified in 1988 by Modic and colleagues: Modic 1 changes correspond to inflammatory signal of the vertebral endplates, Modic 2 to fatty signal and Modic 3 to fibrous signal.4 5
As early as 1990, Revel and colleagues,6 on the basis of X-ray findings, reported that rapid intervertebral space narrowing ≥50% in less than 2 years, in the absence of specific causes of disc disease, was associated with inflammatory-like LBP, which responded better to non-steroidal anti-inflammatory drugs than did other causes of non-specific LBP. The authors suggested that this imaging and clinical phenotype could be related to an ‘active’ process of discolysis and named it ‘rapidly destructive intervertebral disc disease’.6 They further related this condition to Modic 1 changes.7 8 They also observed that the condition was associated with increased serum levels of inflammatory markers such as highly sensitive C-reactive protein9 and therefore might reflect an ‘active discopathy’.7 The concept of active discopathy now encompasses clinical, biological and imaging features that reflect an ‘activation’ (ie, ‘inflammatory flare’) of a previously unremarkable DDD in a subgroup of patients with chronic LBP.10
Active discopathy shares some similarities with acute flares of peripheral osteoarthritis. Likewise, what triggers disc disease activation and how it self-limits remain unknown. A better understanding of mechanisms underlying disc disease activation and its self-limitation, with a specific focus on adverse biomechanical and biochemical factors, could be of clinical relevance.11 Here, we narratively review current disc-centred biomechanical and biochemical hypotheses of disc disease activation.
What is active discopathy?
Degenerative disc disease
To understand active discopathy, we first describe ‘non-active’ disc disease, or DDD. At the macroscopic level, DDD affects the intervertebral disc and also the whole discovertebral complex.12 It is characterised by a fibrillar structure of the annulus fibrosus, cracks of the nucleus pulposus, thinning and erosions of the cartilaginous vertebral endplates, marginal osteophytosis and loss of disc height.13 14 DDD results from an imbalance in intervertebral disc homeostasis with quantitative and qualitative changes affecting intervertebral disc cells and extracellular matrix as well as decreased water content that impair the biomechanics of the intervertebral disc. At the molecular level, DDD is associated with local inflammatory processes characterised by immunological stimulation, neovascularisation and neoinnervation, accelerated cartilaginous and bone remodelling, and changes in lipid and oxidative metabolism.15–18 At the MRI level, the most consistently reported signs of DDD are nucleus pulposus dehydration, decreased disc height, disc bulging or hernia.4 Pfirrmann and colleagues described five MRI grades of DDD.19 The first grade corresponds to a normal intervertebral disc, and higher grades correspond to DDD and have in common a hypointense signal in T2-weighted sequences of a homogenous nucleus pulposus, with no distinction between the annulus fibrosus and nucleus pulposus, and loss of intervertebral disc height.19 However, no consistent anatomoclinical correlation between DDD and LBP has been described.20
Activation of DDD
MRI signs
In the late 1980s, Modic and colleagues described three elementary lesions corresponding to three MRI signal changes affecting the vertebral endplates adjacent to DDD detected on lumbar MRI of 474 patients with LBP5: a Modic 1 signal corresponds to a hypointense signal in T1-weighted MRI sequences and a hyperintense signal in T2-weighted sequences with contrast enhancement after gadolinium injection, indicating local inflammation. Histologically, fissures of the vertebral endplates are observed, with vascularised granulation tissue replacing the normal bone marrow as well as an increased number of osteoblasts and osteoclasts with a thickening of the bony spans, indicating bone remodelling. A Modic 2 signal corresponds to a hyperintense signal in T1 and T2-weighted MRI sequences, indicating fatty replacement of bone marrow. Histologically, cracks in vertebral endplates are observed with granulation tissue but not hypervascularisation. A Modic 3 signal corresponds to a hypointense signal in T1 and T2-weighted MRI sequences, indicating bone sclerosis. The prevalence of Modic changes, whatever their type, ranges from 6% in asymptomatic patients to 43% in patients with LBP.3 Modic changes usually evolve over 1–3 years, from Modic 1 to Modic 2 (then Modic 3), which may reflect a dynamic healing process. However, many longitudinal studies have shown that this evolution is not linear and could be reversed in some cases, with reactivation of the disc disease over time.21–24
Clinical signs
Patients with LBP and active discopathy usually report a flare of previously unremarkable LBP with inflammatory-like features. The influence of DDD activation and radiculopathy incidence has not been studied. First described by Revel and colleagues in the context of rapidly destructive intervertebral disc disease, an inflammatory LBP pattern associated with Modic 1 changes was confirmed by Rannou and colleagues in a cross-sectional study of 36 patients (12 with Modic 1 changes, 12 with Modic 2 changes and 12 with Modic 0 changes). Morning stiffness was longer and more often present in patients with Modic 1 than Modic 0 or 2 changes, and the worst painful moment was during late night and morning in all patients with Modic 1 changes. Discogenic pain was also more frequent in patients with Modic 1 than Modic 0 or 2 changes, including reproduction of the pain during Valsalva manoeuvres and exacerbation of pain in lumbar hyperextension. Despite this inflammatory LBP pattern, patients with LBP and active discopathy do not fulfil criteria for ankylosing spondylitis and criteria sets for inflammatory LBP.25 Other groups further confirmed these clinical findings26 and reported that active discopathy could be associated with poor outcomes of LBP, with low rates of return to work, persistent symptoms at 1 year27 and increased back-specific disability.28 No longitudinal observational studies have assessed the correlations between the evolution of pain and evolution of MRI.
Biological signs
Rannou and colleagues also reported an increase in serum level of high-sensitive C-reactive protein in patients with than without chronic LBP and active discopathy (4.64±3.09 mg/L with Modic 1 changes vs 1.33±0.77 mg/L with Modic 0 changes and 1.75±1.30 mg/L with Modic 2 changes).9 This observation supports that local inflammation occurs at the vertebral endplate level and is consistent with local cell and tissue activation. Time correlations between Modic modification from 1 to 2 and C-reactive protein evolution have not been studied.
Limits of the concept
Even though the concept of active discopathy could now be defined as a syndrome characterised by DDD associated with MRI, clinical and biological signs of activation, its reality and clinical relevance remain controversial among spine researchers29 and its pathogenesis unclear. How DDD activates and how disc disease activation actually self-limits remains largely unanswered. Even though there are some similarities between osteoarthritis and DDD and between inflammatory flare of peripheral osteoarthritis and active discopathy, we agree that the lack of synovial tissue in spine may differentiate the physiopathology of these diseases.
What is the primer for DDD activation?
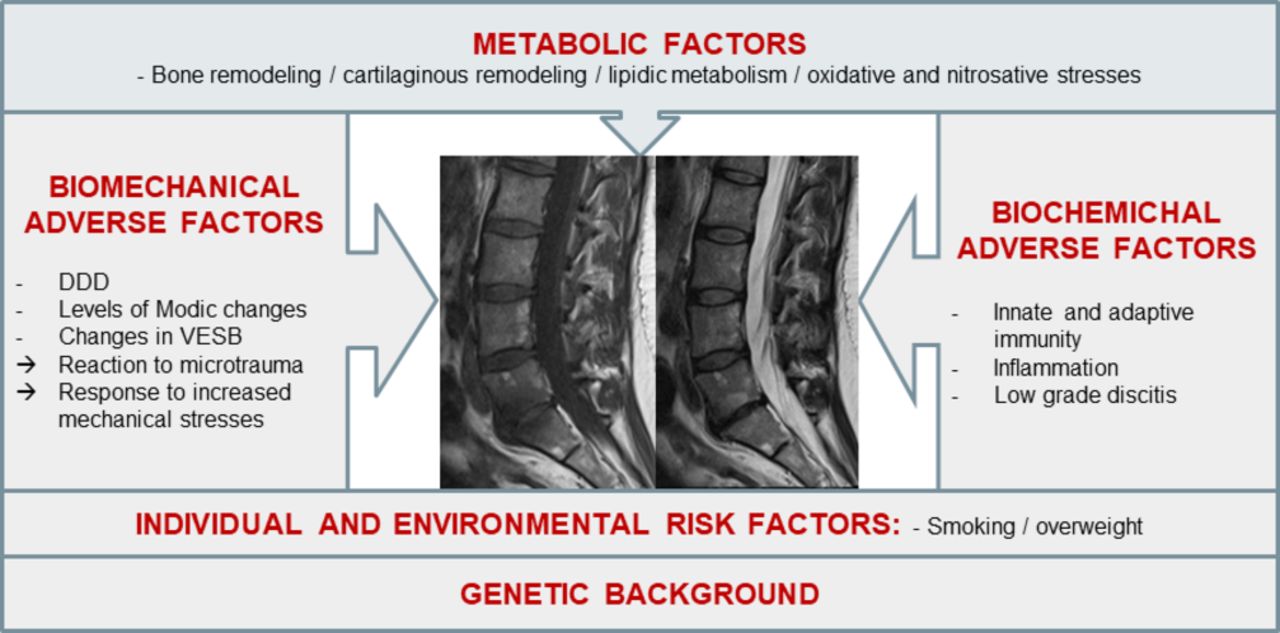
Several factors including adverse biomechanical and biochemical factors may contribute to the activation of DDD, but the onset of active discopathy is most likely multifactorial. Although local inflammation is currently the main therapeutic target, it may be only a late-stage marker of disc disease activation. Other biochemical and metabolic factors, in addition to genetic and biomechanical predisposing factors, could participate in early stages of disc disease activation and be more relevant targets (figure 1).

Main hypothesised aetiopathogenic mechanisms of intervertebral disc disease activation. DDD, degenerative disc disease; VESB, vertebral endplate subchondral bone.
Adverse personal and environmental factors
The association between active discopathy and polymorphisms of various genes including type 1A1, 9A3 and 11A2 collagens; interleukin-1α (IL-1α)30; growth differentiation factor 5; vascular endothelial growth factor; matrix metalloproteinases 3, 9 and 14; a disintegrin and metalloproteinase with thrombospondin motifs 4 and 5; tissue inhibitor of metalloproteinase 3; and vitamin D receptor has been reported.31 In a study of 347 twins, the concordance rates of Modic signal were 0.56 in homozygotic twins versus 0.39 in dizygotic twins. The heritability of Modic changes, whatever their type, was estimated at 30% (16%–43%).32 Other adverse personal factors reported to be associated with active discopathy are smoking, overweight and physical work.33 In an observational cross-sectional study of 100 patients and 500 vertebral levels, Karchevsky and colleagues found Modic changes associated with older age, male gender and increased weight.34 In a retrospective study of 412 patients, Leboeuf-Yde and colleagues reported three independent risk factors for intermittent LBP, DDD or Modic 1 changes on MRI35: smoking ≥20 cigarettes/day, overweight and workload. The risk of a Modic 1 signal was maximum when smoking was associated with physical work (OR=4.6; 95% CI 1.6 to 13.0), then when overweight was associated with physical work (OR=2.9; 95% CI 1.4 to 6.3). Consistently, in 2449 Chinese volunteers, the risk of Modic changes, whatever their type, was increased in smoking and overweight individuals (OR=2.2; 95% CI 1.1 to 4.3). In a recent retrospective study of 16 men and 31 women, Han and colleagues did not confirm the association between smoking and Modic changes but found heavy work and obesity associated more strongly with Modic 3 than other Modic changes.36 Overall, the level of evidence for the above-mentioned clinical associations must be considered low, owing to the cross-sectional or retrospective designs of the studies. No association between other metabolic factors such as diabetes, hypertension, lipid abnormalities and initiation or aggravation of active discopathy has been reported yet.
Adverse biomechanical factors
Inadequate response to mechanical stresses applied to the degenerated intervertebral disc has been suggested to contribute to disc disease activation. DDD results in alterations of the disc biomechanical properties. These biomechanical changes are associated with microfissures or lesions of the vertebral plates adjacent to DDD, increased local expression of proinflammatory molecules such as IL-1β in animal models37 38 and inflammatory and immune responses to the extruded intervertebral disc.39 Consistently, Modic changes are more frequent in L4/L5 and L5/S1 levels than other levels,40 41 which suggests that local biomechanical stress may contribute to the distribution of Modic changes. Albert and Manniche42 found that the prevalence of Modic 1 changes increased from 9% (17/180 discs) to 29% (48/166 discs) after 14 months of follow-up among patients with chronic LBP and that new Modic changes all developed at the level of a herniated disc. The prevalence of Modic changes was increased in patients who had undergone surgery for lumbar disc herniation (OR=3.5; 95% CI 0.8 to 20.8). In a 3-year prospective study of 60 patients, Modic changes, whatever their type, followed disc herniation in 8/13 cases.23 The relation between Modic changes and the evolution of disc herniation was further suggested in a prospective study of 30 patients with herniated disc: after 6 months of conservative treatment, the mean reduction in disc herniation volume was −0.326 and −0.152 cm3 for patients without and with Modic changes.43
Adverse biochemical factors
During non-active discopathy
Whether the degree of DDD is associated with disc disease activation is unclear. Some advanced DDDs are not associated with Modic 1 changes, whereas some Modic 1 changes can occur on vertebral endplates adjacent to early-stage DDD and supposedly mechanically competent intervertebral discs. This observation suggests that besides adverse biomechanical factors, other factors such as adverse biochemical factors may be important contributors to disc disease activation. At the molecular and cellular levels, disc degeneration has been shown to involve several elementary pathways.
Local inflammation with the preponderant role of inflammatory cytokines such as IL-1β and tumour necrosis factor-α promotes cartilaginous extracellular matrix breakdown but also neovascularisation and neoinnervation within the degenerated discs.39 44–48 Innate and adaptive immunity may also participate in intervertebral disc degeneration. The intervertebral disc is the largest avascular organ in the human body and therefore is excluded from immunologic tolerance, contributing to the ‘immune privilege’ of the nucleus pulposus. However, the expression of Fas ligand by nucleus pulposus cells can stimulate apoptosis of activated Fas-positive cytotoxic T lymphocytes. On the one hand, tears of the annulus pulposus will expose the nucleus pulposus to the immune system, which will recognise it as a foreign body and will promote activation of the cytotoxic T lymphocytes and the production of immunoglobulins by activated B lymphocytes. On the other hand, the decreased Fas ligand level in the nucleus pulposus can lead to an unbalanced immune environment during DDD.49 50 In addition, cells of degenerated discs overexpress Toll-like receptors 2 and 4 that can be stimulated by the products of extracellular matrix degradation and amplify the inflammatory and immune response.51 52
Changes in oxidative stress also occur during disc degeneration. Because of its avascular structure, the intervertebral disc is nourished by imbibition from the adjacent cartilaginous and osseous vertebral endplates. Its metabolism is anaerobic and leads to the production of lactates. Local pH is physiologically acidic, between 7.1 for healthy discs and 6.5 or lower for DDD. Acidification of the disc medium is associated with increased apoptosis of disc cells, mainly mediated by abnormal influx of intracellular calcium, and with changes in the disc cell phenotype with increased expression of proinflammatory cytokines such as IL-1β and IL-6 and factors involved in nociception such as nerve growth and brain-derived neurotrophic factors.53 54 In 1969, using a specially constructed antimony pH-electrode needle type, Nachemson reported a negative correlation between pH and disc degeneration, preoperative pain, and the amount of connective tissue reaction around the nerve root in 40 discs from 30 patients who underwent lumbar surgery for ‘rhizopathy’, which suggested that acidification could be a biomarker of painful DDD.55 Dimozi and colleagues found that H2O2-induced stress on disc cells could promote the upregulation of extracellular matrix-degrading enzymes such as matrix metalloproteinases 1, 2 and 9 and a disintegrin and metalloproteinase with thrombospondin motif 5 and the downregulation of tissue inhibitors of metalloproteinase and aggrecan, the major component of the nucleus pulposus.56
Changes in lipid metabolism may also contribute to disc degeneration. At the systemic level, hyperlipidaemia promotes lipid peroxidation and the formation of advanced glycation end-products which bind to extracellular matrix proteins, thereby decreasing its water content. Indirectly, advanced glycation end-products can also stimulate the extracellular matrix degradation by binding to their specific receptor that is expressed by the nucleus pulposus cells of the degenerated disc. This binding can lead to an activation of the NLRP3 inflammasome.57 Some studies have suggested an association between body mass index and Modic changes. However, none examined the relation between fat distribution and Modic changes. Using MRI and dual energy X-ray absorptiometry in 57 patients with or without LBP, Teichtahl and colleagues found Modic changes associated with increased fat mass index (OR=1.20, 95% CI 1.01 to 1.43). Risk of Modic changes was reduced with gynoid fat (OR=0.62, 95% CI 0.43 to 0.89) but increased with android fat (OR=2.11, 95% CI 1.18 to 3.76), which suggests that Modic changes may be associated with a metabolic component.58
Changes in cartilage remodelling involving the cartilaginous vertebral endplates59 and chondrocyte-like cells of the intervertebral disc have been described during DDD.60 During disc degeneration, phenotypical qualitative and quantitative changes affect cartilaginous vertebral endplates and intervertebral disc cell populations, towards an imbalance between extracellular matrix anabolism and catabolism, with an increased expression of matrix metalloproteinases61; decreased expression of type II collagen; abnormal expression of type I, III and X collagens; and decreased expression of Sox-9, the key transcription factor of chondrogenesis.62 Consistently, increased urinary levels of cartilaginous matrix products of degradation such as type II C-telopeptide are associated with the degree of DDD detected on X-ray.63 A decrease in other extracellular matrix proteins such as aggrecan, versican, biglycan, decorin and fibromodulin in both the nucleus pulposus and annulus fibrosus has also been reported.64
During active discopathy
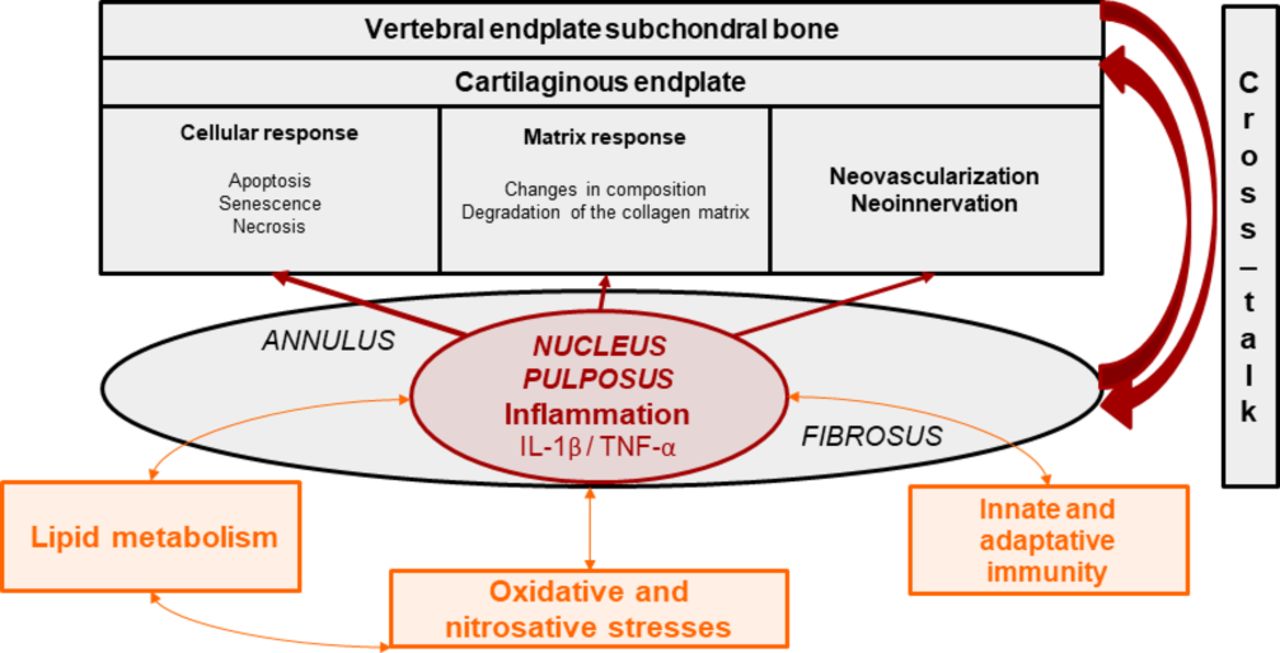
Even though these above-mentioned elementary pathways can concomitantly be activated during disc degeneration, most DDDs remain non-active. This observation suggests that there may be a continuum between non-active and active discopathies that may rely on individual genetically and environmentally conditioned thresholds separating physiological and pathological pathway activation (figure 2).
Revisiting the concept of internal disc disruption: ‘the nuclear theory’ or nucleus pulposus as the primer of local cellular and tissular activation. NSAID, non-steroidal anti-inflammatory drug.
The local expression of proinflammatory cytokines, such as tumour necrosis factor-α, is increased in intervertebral discs with than without Modic 1 or 2 changes. Consistently, Tang and colleagues65 reported increased expression of some inflammasome components such as NACHT, LRR and PYD domain-containing protein-3, caspase-1 and IL-1β in the cartilaginous vertebral endplates. Consistently with Crock66’s concept of ‘internal disc disruption’,67 Ma and colleagues developed the hypothesis that the nucleus pulposus, in the absence of normal vascularisation under physiological circumstances, can be considered an immune sanctuary. When presented to immune cells of adjacent vertebral plates that will recognise them as powerful antigens, nucleus pulposus components trigger an autoimmune and inflammatory cascade and promote subsequent inflammation, neovascularisation and neoinnervation involving adjacent vertebral endplate subchondral bone, detectable as Modic 1 changes on MRI.68
Only one study specifically assessed the link between oxidative stress and active discopathy. Belge Kurutas and colleagues reported an increase in oxidative and nitrosative stress markers with an increase in serum levels of nitric acid, 3-nitrotyrosine and malondialdehyde and a decrease in catalase and superoxide dismutase activities in 10 patients with Modic 1 changes as compared with 12 and 10 patients with Modic 2 and 3 changes, respectively.69
Some observations have implicated lipid metabolism in disc disease activation. Vertebral bone marrow is rich in adipose tissue in elderly men and at the lower lumbar level. Bone marrow adipose tissue is composed of unsaturated fatty acids and low-density oxidised lipoproteins, capable of activating Toll-like receptors 2 and 4 and maintaining and amplifying inflammatory and immune reactions observed in active discopathy.70 71
Changes in cartilage remodelling have also been described during active discopathy and include decreased expression of chondrogenesis transcription factors such as type 2 collagen and Sox-9.65 Cevei and colleagues reported changes in collagen populations depending on Modic 1, 2 or 3 changes, with a decrease in type 1 and 2 collagen levels and an increase in type 3, 4 and 5 levels with Modic 2 changes and a decrease in levels of all collagens and a quasidisappearance of proteoglycan level with Modic 3 changes.14
Increased local bone remodelling has also been associated with Modic 1 changes. Using histomorphometric analyses, Perilli and colleagues showed Modic 1 changes associated with greater bone remodelling, Modic 2 changes with decreased remodelling and increased bone formation and Modic 3 changes with osteosclerosis with increased bone formation and decreased bone resorption.72 Briggs and colleagues reported no differences in bone mineral density between 11 patients with LBP and Modic 1 changes and 10 healthy controls.73 Intervertebral discs and vertebral endplate bone marrow associated with Modic changes express pro-osteoclastic factors and neurotrophic receptors.74 Torkki and colleagues also found increased expression of cytokines involved in osteoclast differentiation and proliferation in intervertebral discs with Modic 1 and 2 changes versus Modic 0 changes.75 In a cross-sectional study of 101 patients with lumbar Modic 1 changes, Nguyen and colleagues found in young men that Modic 1 changes were more frequent at L5/S1 level and associated with mild DDD whereas in older women Modic 1 changes were more frequent at the L4/L5 level and associated with advanced DDD, which indicates perhaps a greater involvement of bone remodelling in disc degeneration associated with active discopathy.41
Finally, some authors suggested that disc disease activation could have an infectious cause, namely discitis with anaerobic germs such Propionibacterium acnes.76 77 However, this hypothesis has not been confirmed by independent groups.78 79
What are the clinical and therapeutic implications of the concept of active discopathy?
Clinical implications
The clinical diagnosis of active discopathy is now facilitated by a comprehensive description of clinical and imaging signs associated with this condition, namely a flare of previously unremarkable chronic LBP with discogenic and inflammatory-like features and Modic 1 changes on MRI. When isolated, these clinical and imaging signs lack specificity. The epidemiological association between active discopathy and adverse personal and environmental factors suggests increased risk of developing an active discopathy for some patients with chronic LBP, who therefore may benefit from a specific treatment and follow-up. Most consistently reported adverse personal and environmental factors are overweight, smoking and physical work, but the overall level of evidence is low due to the methodological weakness of the studies. Other important adverse factors may not have been addressed yet.
Finally, LBP is only a symptom. As for any chronic painful condition, patients with chronic LBP may have more than one cause of pain. Even though active discopathy may explain the acute inflammatory component of LBP, it does not address other contributors to chronic pain such as psychological distress, catastrophism, fear-avoidance beliefs, job dissatisfaction, work absenteeism and low educational level. As for any patient with chronic LBP, these factors should be finely assessed, and modifiable factors concomitantly treated.
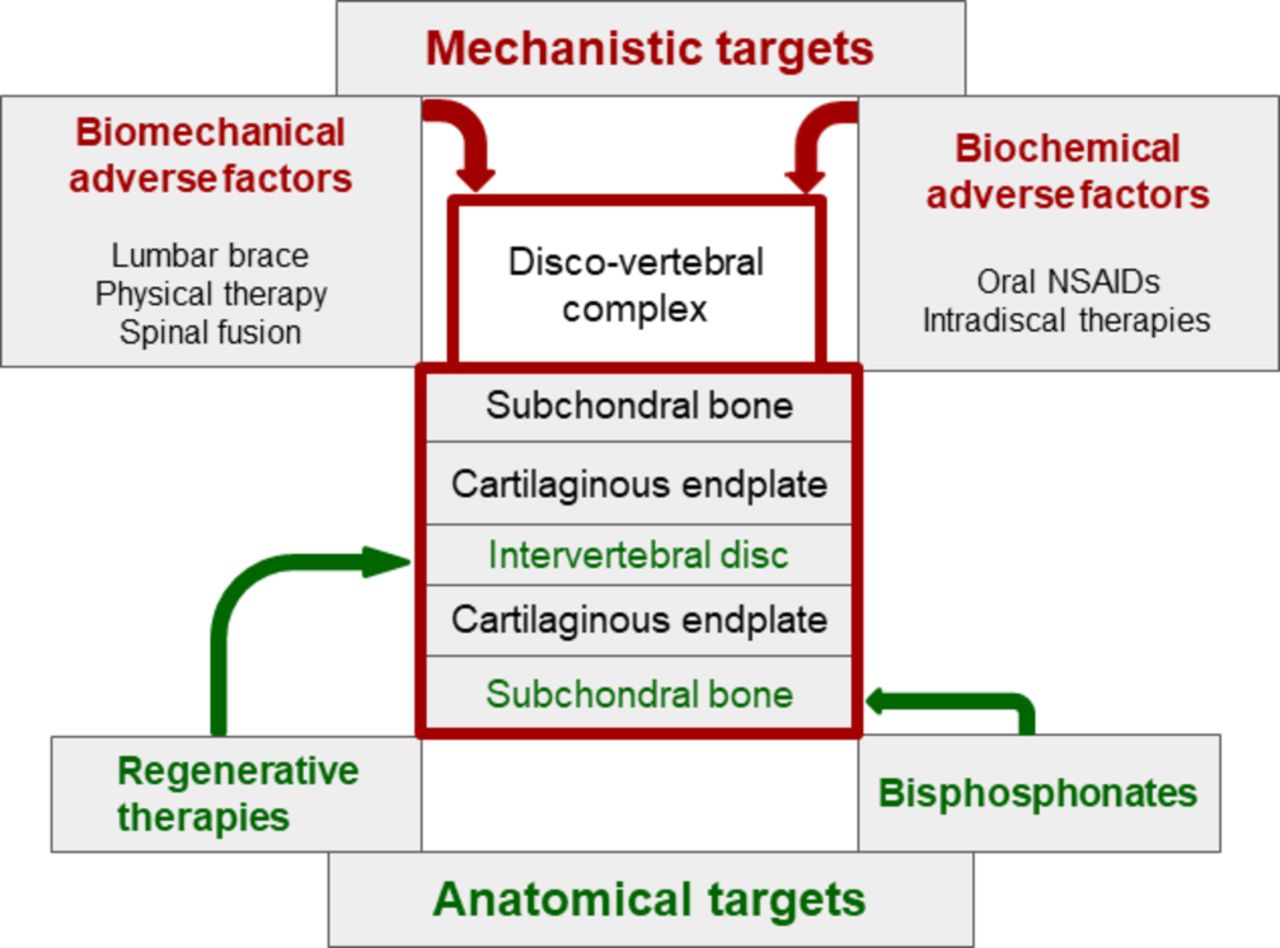
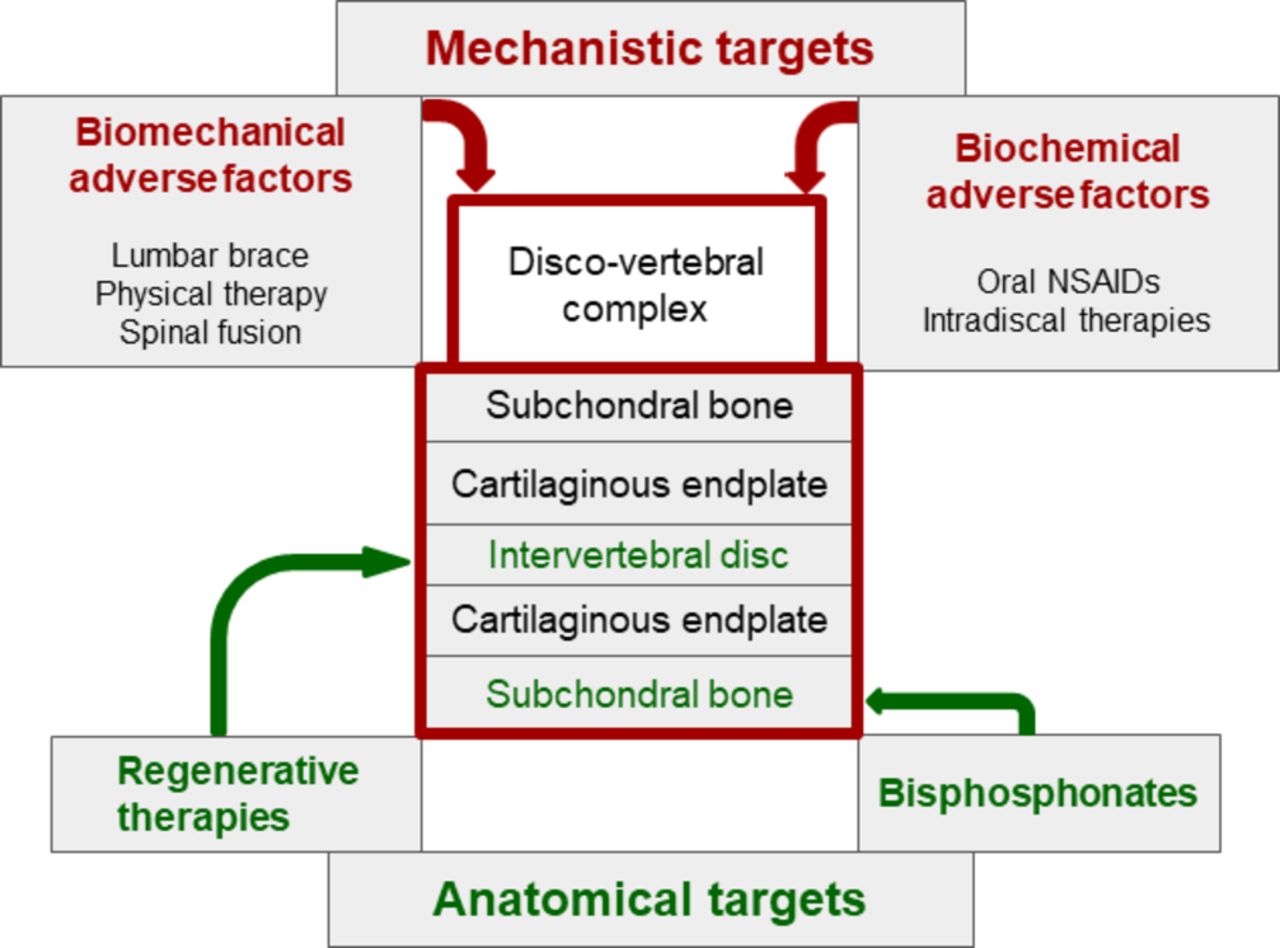
Therapeutic implications (figure 3)

{kind=link}
{kind=link}
{kind=link}
Towards targeted non-pharmacological and pharmacological treatments in patients with chronic low back pain and active discopathy. IL, interleukin; TNF, tumour necrosis factor.
As previously described, active discopathy is a multifactorial disease, and different targeted therapeutic approaches have been proposed. Mechanistic approaches aim at combating biomechanical or biochemical adverse factors, whereas structural approaches aim at reversing alterations involving anatomical structures, mostly subchondral bone and intervertebral disc. Both approaches are often combined. However, we have no evidence or practice-based guidelines for the management of active discopathy.
Biomechanical interventions including lumbar bracing,80 physical therapy81 and ultimately lumbar fusion78 and biochemical systemic or local interventions including intravenous biphosphonates82–84 or intradiscal therapies with glucocorticoids,22 85 86 tumour necrosis factor-α inhibitor87 or IL-6 inhibitor88 have been offered. To date, only intradiscal injection of glucocorticoids has shown a short-term but clear clinical benefit on pain in a high-level randomised controlled trial of 135 patients with chronic LBP and active discopathy.11 Physical activity as a specific treatment for patients with chronic LBP and active discopathy has not been assessed. Studies assessing the long-term effects of this type of treatment are needed. In addition, because chronic LBP is only a symptom and concomitant causes may be involved, a multidisciplinary approach is often necessary to obtain sustained positive effects of treatments.
In the past decades, regenerative medicine of the intervertebral disc has raised intense interest. It has involved intradiscal injection of growth factors, with or without plasma-rich platelets, gene therapy or cell grafting (autologous haematopoietic stem cells, mesenchymal stem cells and autologous chondrocytes) to repair the intervertebral disc.89–91 The RESPINE project (European Horizon 2020 project ID 732163: REgenerative therapy of intervertebral disc: a double blind phase 2b trial of intradiscal injection of mesenchymal stromal cells in degenerative disc disease of the lumbar SPINE unresponsive to conventional therapy) will assess, via a multicentre, randomised, controlled, phase 2b clinical trial including 112 patients with DDD, the efficacy of an allogenic intervertebral mesenchymal stem cell-based therapy. However, as previously stated, active discopathy is a whole-organ disease, involving the intervertebral disc and also surrounding anatomical structures. Therefore, the interest of regenerative medicine targeting only the intervertebral disc may be limited. In addition, the benefits of regenerative medicine approaches have not been proven in high-level clinical trials.
Conclusions and perspectives
DDD is frequent and usually asymptomatic, whereas Modic 1 changes involving the vertebral endplates adjacent to a DDD are associated with inflammatory-like chronic LBP and low-grade local and systemic inflammation, giving rise to the concept of active discopathy. A better understanding of mechanisms underlying disc disease activation and its self-limitation seems of clinical relevance because it could lead to designing more targeted pharmacological and non-pharmacological interventions in the subgroup of patients with chronic LBP and active discopathy (box 1).
Research agenda
1. Improving the phenotyping of patients with chronic low back pain and active discopathy:
Biological phenotyping: serum biomarkers.
Imaging phenotyping: radiological biomarkers.
Development and validation of classification criteria.
2. Unravelling the pathogenesis of active discopathy activation:
In vivo and in vitro modelling of intervertebral disc disease activation.
3. Developing non-pharmacological and pharmacological treatments more efficiently targeting adverse biomechanical, biochemical and environmental factors and taking into account personomics:
Tailored to phenotype and predominant adverse factors.
Accounting for powerful interactions with adverse social and psychological factors.
Aiming to obtain sustained positive effects on pain, function, patient global assessment and returning to work.
Acknowledgments
The authors thank Mrs Laura Smales for professional copyediting.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
Footnotes
Contributors Conception and design of the study: MB, FR, CN. Drafting of the original protocol: MB, FR, CN. Coordination of the study: CN. Acquisition of data: MB, CN. Obtained funding: CN. Drafting of the present manuscript: MB, CN. Final approval: MB, MMLC, FR, CN.
Funding This study was funded by Assistance Public-Hôpitaux de Paris (Project No MERRI-AAP-2017-008).
Competing interests None declared.
Patient consent Not required.
Provenance and peer review Commissioned; externally peer reviewed.
Data sharing statement No additional data are available.