Article Text

Abstract
Objective To test whether daily high-dose vitamin D improves recovery after unilateral total knee replacement.
Methods Data come from a 24-month randomised, double-blind clinical trial. Adults aged 60 and older undergoing unilateral joint replacement due to severe knee osteoarthritis were 6–8 weeks after surgery randomly assigned to receive daily high-dose (2000 IU) or standard-dose (800 IU) vitamin D3. The primary endpoints were symptoms (Western Ontario and McMaster Universities Arthritis Index pain and function scores) assessed at baseline, 6, 12, 18 and 24 months in both knees, and the rate of falls over 24 months. The secondary outcomes were sit-to-stand performance, gait speed, physical activity and radiographic progression in the contralateral knee.
Results We recruited 273 participants, 137 were randomised to receive 2000 IU and 136 were randomised to receive 800 IU vitamin D per day. 2000 IU vitamin D increased 25-hydroxyvitamin D levels to 45.6 ng/mL and 800 IU vitamin D to 37.1 ng/mL at month 24 (p<0.0001). While symptoms improved significantly in the operated knee and remained stable in the contralateral knee over time, none of the primary or secondary endpoints differed by treatment group over time. The rate of falls over 24 months was 1.05 with 2000 IU and 1.07 with 800 IU (p=0.84). 30.5% of participants in the 2000 IU and 31.3% of participants in the 800 IU group had radiographic progression in the contralateral knee over 24 months (p=0.88).
Conclusions Our findings suggest that a 24-month treatment with daily 2000 IU vitamin D did not show greater benefits or harm than a daily standard dose of 800 IU among older adults undergoing unilateral total knee replacement.
- knee osteoarthritis
- orthopaedicsurgery
- rehabilitation
This is an Open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Epidemiological studies suggest that increased vitamin D intake and higher 25-hydroxyvitamin D levels may prevent structural progression of knee osteoarthritis (OA).
However, all four recent clinical trials testing vitamin D supplementation found no such benefit among patients with early to moderate OA not yet at the stage of surgery.
What does this study add?
In this trial, we focus on older adults aged 60 years and older undergoing unilateral total knee replacement due to severe knee OA and include the risk of falling as a primary endpoint.
Further, we compare a standard of care dose of vitamin D (800 IU per day) with a high dose (2000 IU vitamin D) to explore if a higher dose is more effective.
For OA outcomes, we assess pain and function both for the operated and the contralateral knee.
While participants showed significant improvements in symptoms in the operated knee and no worsening of symptoms in the contralateral knee, a daily higher dose of 2000 IU did not show greater benefits than a daily standard dose of 800 IU; this included a similar rate of falling between treatment groups and a similar per cent of participants with radiographic progression in the contralateral knee over 24 months.
How might this impact on clinical practice?
Our findings suggest that a daily dose of vitamin D greater than 800 IU vitamin D is not needed for recovery after unilateral total knee replacement due to severe knee OA.
The efficacy of an 800 IU dose without a pure control group could not be evaluated.
Our findings also suggest that the higher daily dose of 2000 IU compared with 800 IU vitamin D per day is safe among older adults undergoing unilateral knee replacement due to OA.
Introduction
Osteoarthritis (OA) is the leading cause of disability in later age,1 and approximately 30% of individuals aged 65 and older today have symptomatic OA marked by pain in the affected joint.2 3 Despite its frequency, OA is a condition that is poorly understood, with no specific treatments to prevent or reverse the condition.4 5 Therefore current interventions in patients with OA are limited to symptomatic pain relief. This is initially achieved by the use of pain medication, and later with total joint replacement.5–7 However, joint replacement does not fully restore function in most individuals.8 9 Further, adults aged 65 and older have a more than 50% chance of the disease on the contralateral joint,10–12 which often progresses rapidly10 and likely impacts negatively on recovery after total joint replacement.
One promising secondary prevention strategy in older individuals with symptomatic knee OA may be oral vitamin D supplementation. Epidemiological studies suggest that increased vitamin D intake and higher 25-hydroxyvitamin D (25(OH)D) levels may be a strategy to prevent structural progression of knee OA.13 14 The potential benefit has been attributed to a direct effect of vitamin D on cartilage cells based on preliminary studies and in vitro findings.15–17 However, all three recent clinical trials that assessed radiographic progression among patients with symptomatic knee OA found no benefit on MRI-based tibial cartilage volume18 19 or X-ray-based joint space narrowing20 with vitamin D supplementation compared with placebo.
Alternatively, vitamin D may have a beneficial effect on muscle surrounding the OA-affected joint.21 This is supported by the presence of the vitamin D receptor,22–24 as well as an evidence from several clinical trials suggesting that daily 800 IU vitamin D supplementation among adults aged 65+ at risk of vitamin D deficiency improves lower extremity function25 and reduces the risk of falls26 and related fractures.27 28 Given that vitamin D deficiency is common among individuals with knee OA29 and symptomatic knee OA has been shown to double the risk of falls and hip fractures,30 vitamin D supplementation may have clinical relevance in these patients. Notably, one smaller clinical trial of 107 patients suggested a significant improvement in both pain and function among middle-aged adults treated with vitamin D compared with placebo.31
To our knowledge, no clinical trial has been performed to test the benefit of vitamin D on fall prevention or recovery after unilateral knee replacement among patients with severe knee OA. Therefore, our primary goals were to examine whether 2000 IU vitamin D improved symptoms (pain and function) in the operated knee, reduced falls and slowed the progression of symptoms in the contralateral knee, relative to a standard dose of 800 IU vitamin D. Notably, the study did not include a placebo group as the ethical commission at the time requested that the comparator should be standard-dose 800 IU vitamin D to allow compliance with current recommendations given the high risk of falls and hip fractures among seniors with knee OA.30 The high dose of 2000 IU vitamin D was chosen because it was previously found to be more effective than the standard 800 IU vitamin D in shifting most individuals to a target therapeutic range of 24–30 ng/mL,32 where a maximum benefit with regard to knee OA disease progression33 as well as fall and fracture prevention was expected.28 34
Methods
Trial design
This is a single-centre, double-blind randomised trial. All participants gave written informed consent for participation in the trial. The trial was registered at ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/results/NCT00599807. Clinical visits were performed by all participants at baseline (6–8 weeks after unilateral total knee replacement surgery), and at months 6, 12, 18 and 24 after baseline. Between clinical visits, the study nurses called the participants every 2 months to assess falls, adverse events and adherence to study medication.
Setting and locations
The study was performed at the Center on Aging and Mobility at the University Hospital Zurich, with the first patient enrolled in the study in January 2008 and the last patient follow-up visit in March 2014.
Participants and screening procedures
Participants were recruited from two large hospital centres (Schulthess Clinic Zurich, Triemli City Hospital) with a multistep screening process (see also figure 1). Those who were eligible based on the prescreening questionnaire and gave their preconsent to participate in the trial filled out the Western Ontario and McMaster Universities Arthritis Index (WOMAC; score 0–100) prior to their surgery. This instrument was used to capture presurgery symptoms, and later to stratify randomisation. Those who met all the inclusion and exclusion criteria and gave written informed consent (n=273) were enrolled in the trial at 6–8 weeks after surgery.
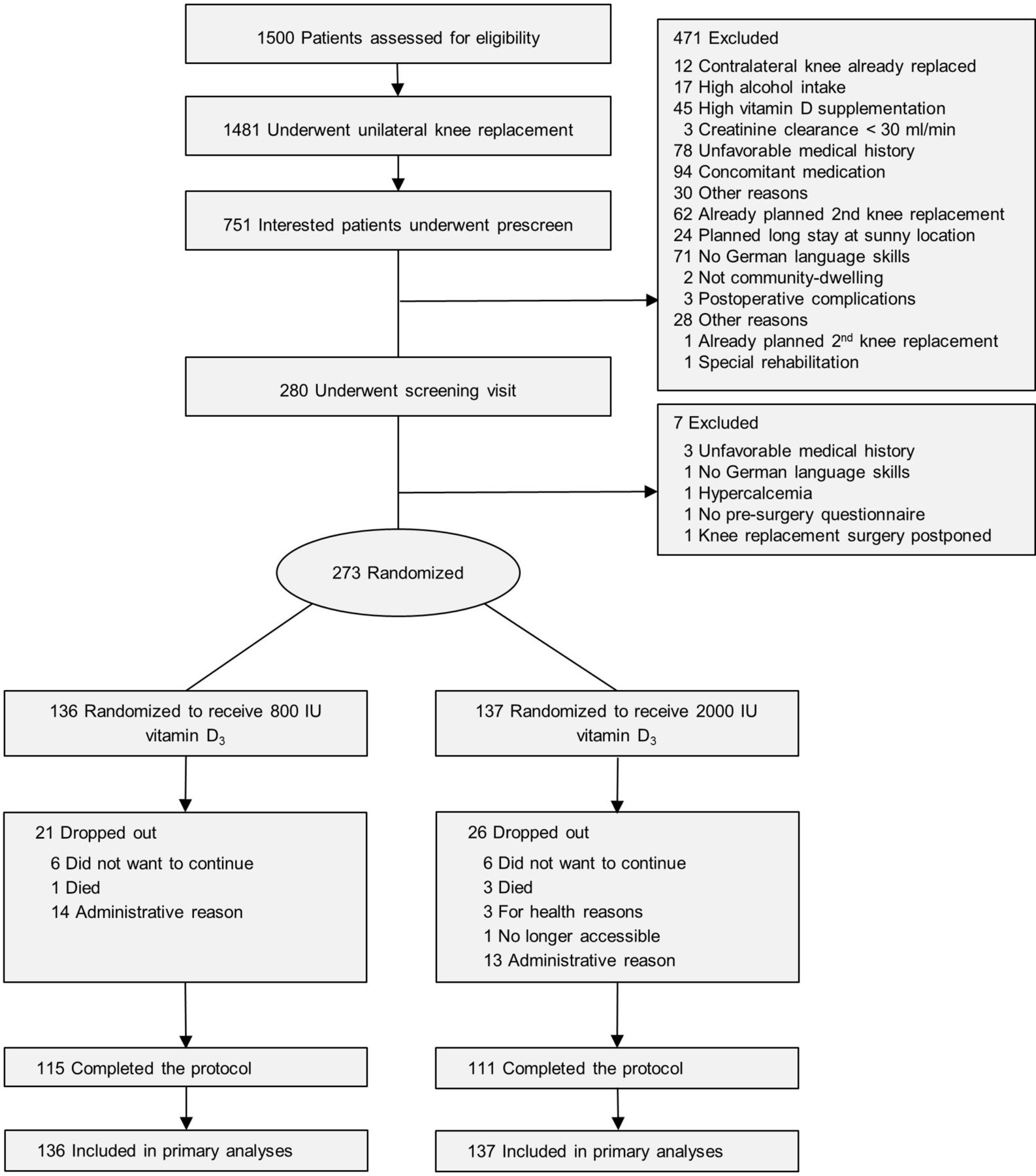
Study recruitment and follow-up.
Inclusion and exclusion criteria
Eligible patients were aged 60 years and older, underwent unilateral total knee replacement due to severe knee OA and did not plan bilateral knee replacement within the next 2 years. Further eligibility criteria included willingness to stop current vitamin D and calcium supplements during the trial, fluent oral and written language skills in German, and a score of at least 24 (of 30) points in the Mini-Mental State Examination cognitive test. Key exclusion criteria were history of inflammatory arthritis and inability to walk at least 3 m with or without a walking aid (for additional exclusion criteria, see online supplementary appendix 1).
Supplemental material
Interventions
Participants were randomised to either one capsule containing 2000 IU vitamin D3/day or one capsule containing 800 IU vitamin D3/day. Vitamin D capsules were produced by the cantonal pharmacy of Zurich and had identical appearances and taste, and assays confirmed the expected contents (eight assessments of individual batches varied between 2017 and 2115 IU for the high-dose group, and between 797 and 864 IU for the standard-dose group). All participants received a 500 mg supplement of calcium per day (calcium carbonate; Sandoz Switzerland).
Outcomes
Primary outcomes
WOMAC symptoms were assessed for the operated and the contralateral knee (function and pain subscales range from 0 to 100, with 0 indicating optimal function and no pain).35 The rate of falls was assessed over 24 months by diary and by phone calls every 2 months. Falls were defined as ‘unintentionally coming to rest on the ground, floor, or other lower level’; coming to rest against furniture or a wall was not counted.36 The primary endpoints, WOMAC pain and function scores, were assessed at baseline, 6, 12, 18 and 24 months in both knees, and the rate of falls over 24 months.
Secondary outcomes
Repeated sit-to-stand test performance and 4 m normal gait speed were assessed at all clinical visits. Physical activity was measured at baseline, 12 and 24 months by a seven consecutive day ankle-worn ambulatory activity monitor (StepWatch Step Activity Monitor, Cyma, Seattle, Washington)37 38 (see online supplementary appendix 1 for additional details on the physical activity assessment and analysis). Radiographic progression in the contralateral knee was assessed at baseline and at 24-month follow-up (see section above) and is detailed in online supplementary appendix 1. In short, we used the Multicentre Osteoarthritis Study (MOST) study centre standardised X-ray assessment procedures,39 and X-rays were evaluated by two blinded expert readers using two published approaches detecting changes in radiographs due to OA.40 41
Randomisation–blinding–treatment allocation
Randomisation lists were computer-generated by the trial statistician (EJO), with separate lists stratified by presurgery WOMAC function score (<52 vs ≥5235 in the operated knee, presence of OA in the non-operated knee and recruitment centre). Each list was blocked in groups of 4 to assure balance. The randomisation lists were sent directly and exclusively to the cantonal pharmacy in Zurich, Switzerland, which carried out the blinding and labelling of the study medication. This pharmacy was located outside the study centre with no access for study team members. The participants and all trial staff were blinded to treatment allocation.
Vitamin D and other laboratory assessments
Fasting blood and urine (second voiding) samples were taken between 08:00 and 09:30 at all clinical visits. The 25(OH)D serum concentrations were measured by a sensitive high-performance liquid chromatography mass spectrometry with multiple steps of mass spectrometry selection (HPLC-MS/MS) method42 43 included in the National Institute of Standards and Technology/National Institutes of Health Vitamin D Metabolites Quality Assurance Program44 at baseline, 6 months and 24 months. Intact parathyroid hormone (iPTH), serum calcium and creatinine, and urinary calcium to creatinine ratio were measured at baseline and at months 6, 12, 18 and 24 using the cobas 8000 system and assays from Roche Diagnostics (Rotkreuz, Switzerland) c501 machine.
Sample size analysis
Our study was designed to enrol 287 patients in order to detect a 38% relative difference in the rate of falls between the two intervention groups. For the rate of falls, our power calculation is based on a conservative estimate from a European trial data on ambulatory older women, which suggested a 46% difference in the rate of falls between the vitamin D group and the control group over a 12-month follow-up.45 We reduced the expected rate difference to 38% as our trial included men and women.46 Assuming full 24-month follow-up on 220 of the patients and at least 12-month follow-up on 234 of the patients, we would have had a power of 80% (two-sided alpha=0.05). During the trial, we recruited slightly fewer patients (273), but more of them (226) completed the full 24-month protocol, providing adequate power for our analyses.
Statistical analyses
A longitudinal linear regression with random intercepts for each patient was used to account for correlation over time (baseline, 6, 12, 18 and 24 months) for continuous variables. The primary predictors in each model were categorical time, intervention group, and the interaction between intervention and time. These models were run initially with no adjustment covariates. The models for primary and secondary outcomes adjusted for design stratification covariates (preoperative WOMAC function score (<52 vs ≥52) in the operated knee, presence of OA in the contralateral knee and recruitment centre) as well as four prechosen covariates considered to be clinically important (baseline age, gender, body mass index (BMI) and Charlson Comorbidity Index). Since five primary outcomes were specified in our protocol (pain and function over time in the operated and contralateral knee and rate of falls), a Bonferroni-adjusted p value of <0.01 should be considered evidence of significance. For the rate of falls, we used a simple Poisson regression model, including the number of months of follow-up as an offset. For the percentages of patients with any falls, we used a logistic regression model and included a patient’s follow-up time as a covariate in all models.
Each of the analyses was an intention-to-treat analysis. Predefined subgroup analyses included gender, preoperative WOMAC function score (<52 vs ≥5235 in the operated knee and presence of OA in the contralateral knee). We first added a three-way interaction term between each of these characteristics, time and intervention group to the model. If that term was significant at p<0.10, we examined the intervention effect within each subgroup.
Results
Participants
Figure 1 shows the conduct of the trial. Because of slow accrual, the study closed before the last 27 subjects reached their 24-month follow-up visits. These participants were included in all analyses. Notably, of the actual 20 dropouts, 4 were deaths, and thus the true dropout rate was 5.8% (16 of 273). At baseline, all variables but weight and BMI were balanced between the treatment groups (table 1).
Baseline characteristics by treatment
Adherence to study medication and changes in 25(OH)D and iPTH serum concentrations by treatment groups and biochemical safety
Based on 2-monthly phone-based assessments, 93% of participants in the 800 IU group and 92% in the 2000 IU group were at least 80% adherent to the vitamin D study medication. Changes in 25(OH)D serum concentrations are shown in table 2. After adjustment for all covariates, 800 IU vitamin D per day increased 25(OH)D to 40.4 ng/mL at 6 months and to 37.1 ng/mL at 24 months. With 2000 IU vitamin D, 25(OH)D levels increased to 47.0 ng/mL at 6 months and 45.5 ng/mL at 24 months. Adjusted levels for iPTH did not differ by treatment, although there was a significant increase over time in the 800 IU group (table 2).
Change in serum 25(OHD) and iPTH concentration at 6-month and 24-month follow-up by treatment
Primary endpoints: symptoms (pain and function) and falls
Both treatment groups experienced a significant functional improvement and pain reduction from baseline (6–8 weeks after unilateral knee replacement) across time in the operated knee (p<0.0001), while symptoms remained unchanged over time in the contralateral knee (see table 3A and figure 2). In both knees, symptoms did not differ by treatment at any time point, nor over the whole follow-up across 24 months.

{kind=link}
{kind=link}
WOMAC (Western Ontario and McMaster Universities Arthritis Index) symptoms in the operated knee and contralateral knee over time and by treatment. The number of patients followed in the 800 IU group at each time point was baseline (BL)=136, 6 months=130, 12 months=130,18 months=123 and 24 months=115. The number of patients followed in the 2000 IU group at each time point was baseline=137, 6 months=129, 12 months=125, 18 months=118 and 24 months=111. Data points show least-square means based on repeated measurement analyses that included an indicator variable for treatment (800 IU vitamin D vs 2000 IU vitamin D), time, and the interaction between treatment and time, and were adjusted for the preoperative WOMAC function score (<52 vs ≥52) in the operated knee, presence of osteoarthritis in the non-operated knee, hospital site, and baseline age, gender, body mass index and Charlson Comorbidity Index (score 0–37). Symptoms in the operated knee (black) improved significantly over time for both function and pain, but not differentially by treatment. Symptoms in the contralateral knee (grey) stayed constant over time and did not change between treatment groups.
(A) Primary and secondary endpoints — by treatment
Over 24 months 158 of 273 participants sustained 307 falls, of which 157 occurred in the 800 IU group and 150 in the 2000 IU group. Fourteen participants had more than four falls (nine with standard-dose and five with high-dose), which according to protocol were truncated to four for the analyses in table 3A. The adjusted mean number of falls was similar in both treatment groups over the 24 months of follow-up (1.07 falls in the standard vs 1.05 in the high-dose vitamin D group, p=0.84). A similar pattern was seen over the first 12 months of follow-up (see table 3A). Although not a primary endpoint, we also compared the percentage of patient who fell and found no difference between the arms (see table 3A).
Secondary endpoints: lower extremity function, physical activity and radiographic progression in the contralateral knee
Both treatment groups experienced a significant gain in lower extremity function from baseline (6–8 weeks after unilateral knee replacement) to 6, 12, 18 and 24 months of follow-up (p<0.0001; see table 3B). For both lower extremity function test sit-to-stand (STS) and gait speed, the difference between treatment groups approached significance (p=0.09 for each), with a possibly more pronounced improvement in the 2000 IU group. StepWatch-based physical activity also increased significantly over time, but not differentially so by treatment group (p=0.84; see table 3B).
Based on standardised X-rays of the contralateral knee, 31.3% of participants in the standard-dose and 30.5% of participants in the high-dose group progressed from baseline to 24 months, but again with no difference between the treatment groups (p=0.88; see table 3B).
Subgroup analyses
As predefined in the study protocol, we tested three interactions with treatment for the primary endpoints: by gender, by WOMAC function in the operated knee prior to surgery (<52 vs ≥52) and by OA in the contralateral knee at baseline (see online supplementary appendix 2). One interaction with treatment was found for gender, suggesting that high-dose vitamin D may improve WOMAC pain in the operated knee among men but not among women. The other interaction was found for treatment and function prior to surgery, where seniors with a lower function may have an increased chance to fall with high-dose vitamin D.
Biochemical safety
Regarding biochemical safety (see online supplementary appendix 3), mean serum calcium, and creatinine levels and mean urinary calcium excretion did not differ by treatment group at baseline and at 6, 12, 18 and 24 months of follow-up. There were few cases (between 0 and 4) of mild hypercalcaemia (>2.6 mmol/L) balanced between the treatment groups at all time points. At none of the time points were there any cases of overt hypercalcaemia >3.0 mmol/L (see online supplementary appendix 3).
Discussion
Our study shows that among patients aged 60 and above, a higher dose of 2000 IU daily vitamin D supplementation compared with a standard dose of 800 IU daily vitamin D supplementation does not improve outcomes after total unilateral knee replacement. This includes pain and function in the operated and contralateral knees, as well as the rate of falls. While this strongly suggests that a higher dose of 2000 IU is not necessary for recovery after unilateral total knee replacement, we cannot determine the degree of efficacy of the 800 IU dose without a pure control group.
The only significant dose-differential findings were identified in predefined subgroup analyses by gender and by function prior to surgery. Based on these, we cannot exclude the possibility that men may benefit from 2000 IU superior to 800 IU vitamin D with respect to a significantly greater pain reduction in their operated knee. On the other hand, we also cannot exclude the possibility that 2000 IU vitamin D may increase fall risk significantly among patients who are most limited in their WOMAC function at the index knee prior to surgery.
Our trial is the fifth in a recent series of double-blind randomised controlled trials18–20 31 on vitamin D supplementation among patients with symptomatic knee OA. Compared with the prior trials, we targeted somewhat older adults with a mean age of 70 years versus aged 53–64 and not yet at the stage of surgery in the previous four trials. Unique to our trial is also the concept of targeting WOMAC symptoms individually for the operated and the contralateral knee, to investigate both recovery of the operated knee as well as progression in the contralateral non-operated knee.47 For the latter, we anticipated10–12 and confirmed a high 57% prevalence of radiographic OA in the contralateral knee at baseline. However, contrary to our expectations, we did not find rapid disease progression10 in the contralateral knee, both clinically and radiographically. In fact, for symptoms, the majority of participants in both treatment groups remained stable.
One explanation may be that 800 IU vitamin D may have been sufficient to stop symptomatic progression in the contralateral knee. Supporting such a benefit, in three18 19 31 of the four prior trials, a benefit of vitamin D on WOMAC function could not be excluded, and one31 of the four prior trials found a significant improvement in WOMAC pain. However, regarding structural disease progression, none of the prior trials support such a benefit.
We confirm the high risk of falling in this patient group.48 However, the adjusted mean number of falls over 24 months was similar between the treatment groups. Only in the subgroup with most pronounced functional limitations prior to surgery more falls occurred with 2000 IU vitamin D compared with 800 IU vitamin D. This is consistent with one prior trial among 173 patients with hip fracture, where a possible increase of falls with 2000 IU vitamin D/day vs 800 IU/day could not be excluded (+28%; 95% CI −4% to +68%).34
A limitation of our study is the lack of a pure control group, which is why our trial cannot establish a benefit of daily 800 IU vitamin D over placebo. However, at the time of the trial (start in 2007), the safe upper intake of vitamin D was still 2000 IU, and the ethical commission at the time requested that the comparator should be the standard-dose 800 IU vitamin D to allow compliance with current recommendations given the high risk of falls and hip fractures among seniors with knee OA.30 Also, contrary to our expectations,49 only 31.4% of participants were vitamin D-deficient at baseline, and the achieved 25(OH)D levels only differed by 6.6 ng/mL at 6 months and 8.4 ng/mL at 24 months.34
In conclusion, our findings suggest that daily 800 IU vitamin D may be sufficient for recovery after unilateral total knee replacement due to severe knee OA. However, despite the observed improvement in symptoms among participants of both treatment groups in our trial, we cannot determine the degree of efficacy of the 800 IU dose without a pure control group. More research is needed to clarify the potential benefit of vitamin D on OA symptoms.
References
Footnotes
Contributors HAB-F designed the trial, is the guarantor, received funding for the trial, wrote the analysis plan, performed statistical analyses and wrote the first draft of the paper. EJO codesigned the trial, supervised the analyses as the head biostatistician and contributed to the first draft of the paper. AE was the coordinating study MD of the trial, contributed to data cleaning and contributed to the first draft of the paper. BD-H contributed to the design of the trial and contributed to the first draft of the paper. KF cleaned the data set, contributed to the first draft of the Methods and Results sections, and reviewed the final draft of the paper. HBS contributed to the design of the trial, was an advisor on the falls outcome and reviewed the final draft of the paper. RR contributed to the design of the trial, was an advisor on the WOMAC pain and function outcomes, and reviewed the final draft of the paper. JH collaborated on the radiological assessment of knee OA in the trial, advised on the radiological outcome and reviewed the final draft of the paper. AvE advised and performed the laboratory analyses and reviewed the final draft of the paper. GF advised on the analyses and adjudication of falls and reviewed the final draft of the paper. UM cleaned the data set, contributed to the first draft of the Methods section and reviewed the final draft of the paper. TG advised on the participant selection for the trial, supervised recruitment at the largest recruitment site and reviewed the final draft of the paper. PB contributed to the design of the trial, was an advisor on 25-hydroxyvitamin D measurements and reviewed the final draft of the paper. SS advised on the analyses of comorbid conditions and reviewed the final draft of the paper. PC-B contributed to data cleaning, advised on the statistical analyses and reviewed the final draft of the paper. RT contributed to the design of the trial and the first draft of the paper. WCW contributed to the design of the trial and the first draft of the paper. DF contributed to the design of the trial, is guarantor, coordinated the radiological assessment and contributed to the first draft of the paper. All authors reviewed and approved the final draft of the paper.
Funding This project was funded by a Swiss National Science Foundation Professorship (Swiss National Science Foundations Professorship Grant PP00B-114864; Bischoff-Ferrari HA) the Velux Stiftung (Grant Number 441; Bischoff-Ferrari HA) and the Baugarten Foundation Center Grant to the Center on Aging and Mobility at the University of Zurich and University Hospital Zurich (Bischoff-Ferrari HA).
Competing interests None declared.
Patient consent Obtained.
Ethics approval The study protocol was approved by the ethical committee of the Canton of Zurich and the Swiss Agency for Therapeutic Products (Swissmedic, regulatory agency of Switzerland).
Provenance and peer review Not commissioned; externally peer reviewed.