Article Text

Abstract
Objective To conduct subset analyses of SPIRIT-P2 (NCT02349295) to investigate the efficacy and safety of ixekizumab versus placebo in three subgroups of patients with active psoriatic arthritis (PsA) according to the concomitant conventional synthetic disease-modifying antirheumatic drug (cDMARD) received: any background cDMARDs (including methotrexate), background methotrexate only, or none.
Methods Patients were randomised to receive placebo, ixekizumab 80 mg every 4 weeks (IXEQ4W) or every 2 weeks (IXEQ2W). Efficacy and safety were assessed when patients were subdivided according to cDMARD use at baseline. Efficacy was evaluated versus placebo at week 24 by the American College of Rheumatology criteria (ACR20/50), achievement of minimal disease activity (MDA) state, Disease Activity Index for PsA (DAPSA), 28-joint Disease Activity Score using C-reactive protein (DAS28-CRP), Health Assessment Questionnaire-Disability Index and the 36-item Short-Form health survey physical functioning domain.
Results Regardless of background cDMARD status, ACR20, ACR50 and MDA response rates were significantly higher than placebo with IXEQ4W or IXEQ2W treatment. Similarly, significant improvements were observed relative to placebo for DAS28-CRP and DAPSA across subgroups. Physical function also significantly improved relative to placebo with IXEQ4W treatment regardless of background cDMARD status and with IXEQ2W alone. Percentages of reported treatment-emergent adverse events (AEs), serious AEs (including serious infections) and discontinuations due to AEs in each subgroup were comparable to the overall SPIRIT-P2 population.
Conclusion Ixekizumab was efficacious in patients with active PsA and previous tumour necrosis factor inhibitor (TNFi) inadequate response or TNFi intolerance treated with ixekizumab alone or when added to cDMARDs with subgroup safety profiles that were consistent with that observed in the overall SPIRIT-P2 population.
- DMARDs (biologic)
- DMARDs (synthetic)
- methotrexate
- psoriatic arthritis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Clinical trial data generally suggest that biologics (primarily tumour necrosis factor inhibitors (TNFis)) may be used alone or when added to conventional synthetic disease-modifying antirheumatic drugs (cDMARDs) such as methotrexate with similar efficacy in patients with psoriatic arthritis (PsA), but some registry studies identified differences in effectiveness.
Ixekizumab was shown to be efficacious versus placebo either alone or when added to cDMARD therapy in patients with active PsA who were previously naive to biological treatment.
What does this study add?
This post hoc analysis examines the efficacy and safety of ixekizumab in subgroups of patients treated with ixekizumab alone or when added to cDMARD therapy in a patient population with PsA that was exclusively TNFi-experienced (TNFi-inadequate responder or TNFi-intolerant).
How might this impact on clinical practice?
The present analysis indicates that ixekizumab is efficacious versus placebo in active PsA either as monotherapy or when added to cDMARD therapy.
Introduction
Biologic disease-modifying antirheumatic drugs are commonly prescribed in combination with conventional synthetic disease-modifying antirheumatic drugs (cDMARDs) in patients with psoriatic arthritis (PsA). Interestingly, reports of randomised controlled trials (RCTs) have reported no added benefit when combining biological therapy with cDMARDs and consistent efficacy in patients treated with or without background cDMARDs, but registry studies suggest that differences in effectiveness may exist, as highlighted by drug survival results.1–9 Of note, these RCTs investigated efficacy in populations that included all or some biologic-naive patients with PsA, thus no prior studies have investigated the efficacy and safety of biologics alone or when added to cDMARD therapy in an exclusively tumour necrosis factor inhibitor (TNFi)-experienced population. These patients were either TNFi-inadequate responders or TNFi-intolerant and may not either achieve or maintain efficacy with subsequent TNFis thus requiring therapies with different mechanisms of action.10
Ixekizumab is a high-affinity monoclonal antibody that selectively targets interleukin 17A and has been demonstrated to be highly efficacious in the treatment of PsA in biologic-naive and TNFi-inadequate responder or TNFi-intolerant populations (SPIRIT-P1 and SPIRIT-P2, respectively).11–13 In SPIRIT-P2, 51% of patients used background cDMARDs at baseline, but all patients were cDMARD-experienced.13 As is the case for all published PsA RCTs, the designs of both SPIRIT-P1 and SPIRIT-P2 do not permit direct comparisons between subgroups of ixekizumab alone and when added to cDMARD therapy.12 13 Thus, we conducted subset analyses of SPIRIT-P2 to investigate the efficacy and safety of ixekizumab relative to placebo (PBO) in three subgroups of patients according to the concomitant cDMARD received: (1) any background cDMARDs (including methotrexate (MTX)), (2) background MTX only or (3) none.
Methods
Trial design
The study design of SPIRIT-P2 (NCT02349295) was previously reported by Nash and colleagues.13 In brief, SPIRIT-P2 was a randomised, double-blind, PBO-controlled phase 3 study of ixekizumab in TNFi-inadequate responders (insufficient efficacy or secondary loss of response) or TNFi-intolerant patients with active PsA. Patients were stratified by country and previous TNFi inadequate response (to one or two TNFis) or TNFi intolerance and then randomised 1:1:1 to receive subcutaneous injections of ixekizumab 80 mg every 4 weeks (IXEQ4W), ixekizumab 80 mg every 2 weeks (IXEQ2W) or PBO for the double-blind treatment period (0 to 24 weeks). Patients who were randomised to ixekizumab received a 160 mg ixekizumab as starting dose at week 0. At week 16, patients designated as inadequate responders, whether treated with ixekizumab or PBO, were required to add or modify concomitant medications for rescue therapy. Patient receiving ixekizumab maintained their assigned current ixekizumab dose and those receiving PBO were re-randomised 1:1 to receive either IXEQ4W or IXEQ2W at week 16.
Patient population
Patients (aged ≥18 years) were eligible to participate in SPIRIT-P2 if they fulfilled classification criteria for PsA (CASPAR), had ≥3 of 68 tender and ≥3 of 66 swollen joints, with active or documented history of plaque psoriasis, previously had an inadequate response to one or two TNFis or were intolerant to a TNFi. Patients were also required to have been treated with at least one cDMARD (MTX, hydroxychloroquine, leflunomide (LEF) or sulfasalazine (SSZ)). Patients who were on stable doses with no changes within 8 weeks prior to baseline, of cDMARDs (MTX, hydroxychloroquine, LEF, SSZ) or other select medications (topical corticosteroids of weak potency), oral corticosteroids, opiates and/or non-steroidal anti-inflammatory drugs/cyclo-oxygenase-2 inhibitors) continued these medications without any modifications in the treatment regimen during the 24-week double-blind treatment period unless required for safety reasons or due to designation as an inadequate responder at week 16 as mentioned in the study design section. Changes made at week 16 were required to remain constant through week 24 unless required for safety reasons. During the study, maximum cDMARD dosages allowed were MTX 25 mg/week, hydroxychloroquine 400 mg/day, LEF 20 mg/day and SSZ 3 g/day. For safety reasons, simultaneous use of MTX and LEF was not allowed and treatment with more than one cDMARD at study entry was not allowed per study exclusion criteria.
Efficacy and safety assessments
Efficacy and safety assessments were performed at scheduled study visits (weeks 1, 2, 4, 8, 12, 16, 20 and 24) during the double-blind treatment period. In these subset analyses, only results from week 24 are reported in alignment with the time point specified for the primary objective of SPIRIT-P2.
Efficacy was assessed at week 24 using the American College of Rheumatology (ACR) 20/50 responder indices14 and the following measures: proportion of patients achieving minimal disease activity (MDA) state defined by fulfilment of ≥5 of 7 domains (tender joint count (TJC) ≤1, swollen joint count (SJC) ≤1, Psoriasis Area and Severity Index ≤1 or percent body surface area affected ≤3, Patient’s Assessment of Pain Visual Analogue Scale (VAS) ≤15, Patient’s Global Assessment of Disease Activity VAS ≤20, Health Assessment Questionnaire Disability Index (HAQ-DI) ≤0.5 and tender entheseal points ≤1)15 16; change from baseline in Disease Activity Index for PsA (DAPSA) which is the sum of 68-joint TJC, 66-joint SJC, Patient’s Global Assessment of Disease Activity VAS, Patient’s Global Assessment of Pain VAS and C-reactive protein (CRP; mg/dL)17; DAPSA ≤4 (remission)18 19; DAPSA ≤14 (at least low disease activity (LDA)); DAPSA ≤28 (at least moderate disease activity); change from baseline in 28-joint Disease Activity Score using CRP (DAS28-CRP); change from baseline in HAQ-DI supported by the proportion of patients achieving HAQ-DI minimal clinically important differences (MCID) (improvement ≥0.35)12; and the physical functioning (PF) domain of the 36-item Short Form Survey v2 acute version (SF-36).
Safety was assessed at week 24 by evaluating the incidence of adverse events (AEs; total, mild, moderate and severe), serious adverse events (SAEs, including serious infections) and AEs leading to discontinuation.
Statistical analyses
The subgroups consisted of patients treated with PBO or ixekizumab who were concomitantly receiving at baseline: (1) any cDMARDs (including MTX), (2) only MTX or (3) no cDMARDs. Patients using MTX were a subset of patients using cDMARDs. Therefore, interaction tests were performed using only the background cDMARD (including MTX) use and no background therapy subgroups. For categorical variables, proportions (or percentages) were reported. To assess treatment-by-cDMARD use interaction effect, a logistic regression model was used with the following factors: treatment, cDMARD use and the interaction of treatment-by-cDMARD use. The treatment-by-cDMARD use interaction was tested at the significance level of 0.10. Treatment group differences were assessed within each subgroup using Fisher’s exact test. Continuous variables were analysed using an analysis of covariance (ANCOVA) models. To assess treatment-by-cDMARD use interaction effect, an ANCOVA model was used that included treatment, cDMARD use and a treatment-by-cDMARD use interaction as factors and baseline outcome measure as a covariate. Treatment group differences were assessed within each subgroup using an ANCOVA model, which included treatment as a factor and the baseline outcome measure as a covariate. Least squares mean changes from baseline were reported for continuous variables. Missing values were imputed by non-responder imputation and last observation carried forward for categorical and continuous variables, respectively. All comparisons were made to PBO and no statistical comparisons were made between ixekizumab alone and ixekizumab when added to background cDMARD or MTX use. All analyses were post hoc, except for ACR20 and summaries of common treatment-emergent adverse events (TEAEs), which were prespecified.
Results
In SPIRIT-P2, 363 patients were randomised and received either PBO (n=118, 52 receiving cDMARDs (including MTX), 40 receiving MTX), IXEQ4W (n=122, 60 receiving cDMARDs (including MTX), 48 receiving MTX) or IXEQ2W (n=123, 73 receiving cDMARDs (including MTX), 61 receiving MTX).13 Patient baseline characteristics were comparable relative to PBO across subgroups with a few exceptions (table 1).
Baseline patient demographics and patient characteristics when subdivided according to background cDMARD use and treatment regimen
Efficacy outcomes were significantly improved relative to PBO in patients treated with IXE, regardless of background cDMARD or MTX use, with a few exceptions among the DAPSA-related and HAQ-DI-related results. All efficacy results indicated that patients treated with ixekizumab achieved improvements in PsA disease activity after 24 weeks of treatment and the magnitude of the improvements, as measured by several outcome measures, were comparable to those observed in the overall population regardless of background cDMARD use.13 DAPSA responses showed consistent efficacy results with ACR and MDA responses.13
ACR20 (figure 1A), ACR50 (figure 1B) and MDA (figure 1C) response rates at week 24 in patients treated with IXEQ4W or IXEQ2W were significantly higher relative to PBO, whether the patients received ixekizumab alone or when added to background cDMARDs or MTX.

ACR20 (A), ACR50 (B) and MDA (C) response rates at 24 weeks in patients treated with PBO, IXEQ4W or IXEQ2W alone or when added to background cDMARDs or MTX. ACR20 results in the ITT population for the background cDMARD and monotherapy subgroups were previously published by Nash and colleagues.13 Response rates reported with 95% confidence intervals. The treatment-by-cDMARD interaction p-values for treatment by cDMARD use were 0.5115 for ACR20 and 0.2616 for ACR50.**p<0.01 versus PBO; ***p<0.001 versus PBO. ACR20/50, American College of Rheumatology criteria 20%/50% improvement; cDMARD, conventional disease-modifying antirheumatic drugs; IXEQ4W, ixekizumab every 4 weeks; IXEQ2W, ixekizumab every 2 weeks; ITT, intent-to-treat; MDA, minimal disease activity; MTX, methotrexate; n, number of patients; PBO, placebo.
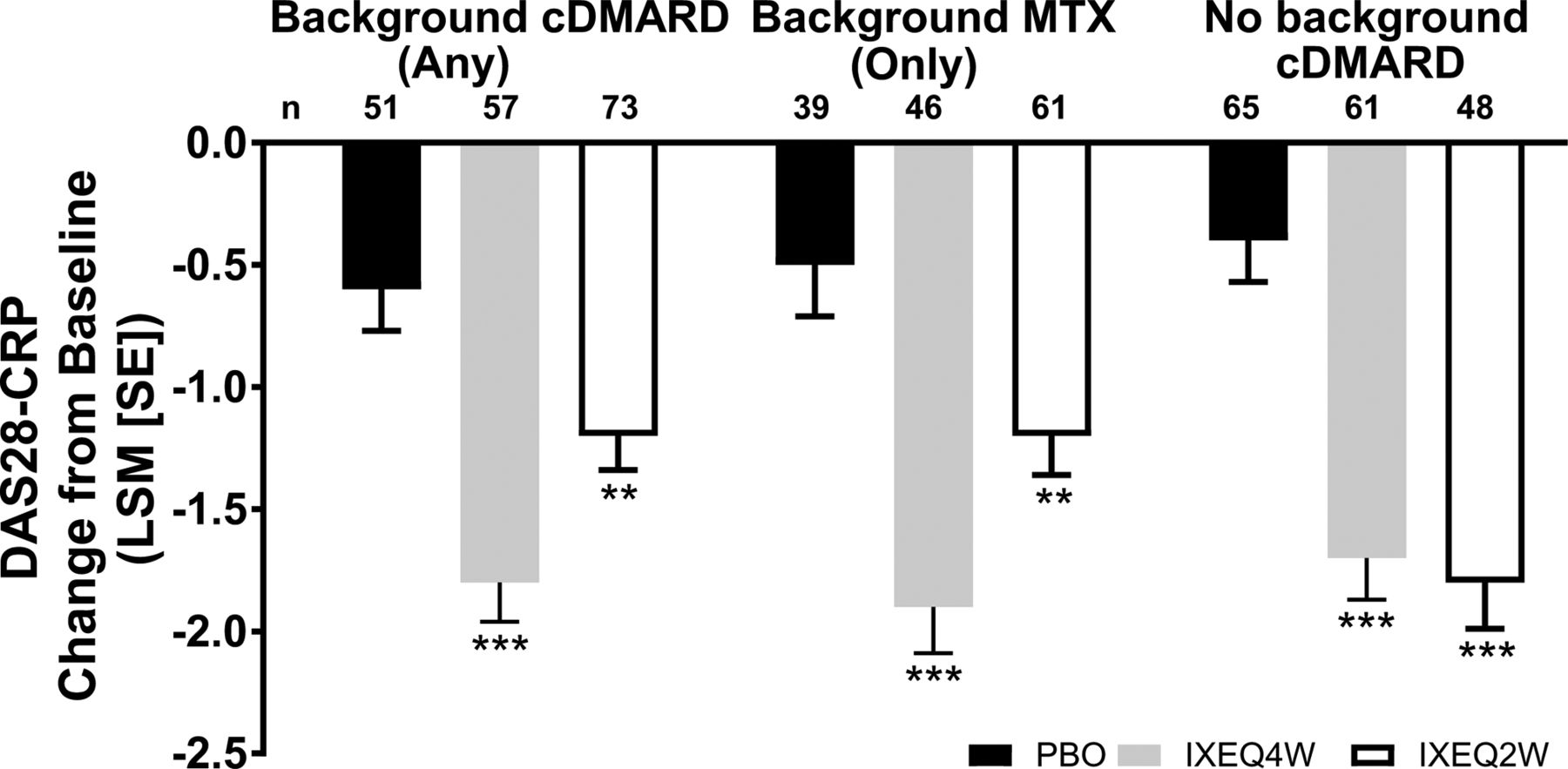
Disease activity, as measured by DAPSA and DAS28-CRP, improved in ixekizumab-treated patients regardless of background cDMARD use at week 24 relative to PBO and reflected the disease activity improvements observed (according to DAS28-CRP) in the overall trial population at week 24.13 IXEQ4W-treated or IXEQ2W-treated patients achieved significant improvements in DAPSA change from baseline relative to PBO at week 24, whether they received ixekizumab alone or when added to background cDMARDs or MTX (figure 2A). Relative to PBO, higher proportions of patients achieved DAPSA ≤28 (figure 2B), DAPSA ≤14 (figure 2C) and DAPSA ≤4 (figure 2D) in all subgroups, and with the exception of DAPSA ≤28 and DAPSA ≤4 (IXEQ2W only), these differences were statistically significant regardless of background cDMARD or MTX use. IXEQ4W-treated or IXEQ2W-treated patients achieved significant improvements in DAS28-CRP change from baseline relative to PBO at week 24, whether they received ixekizumab alone or when added to background cDMARDs or MTX (figure 3).
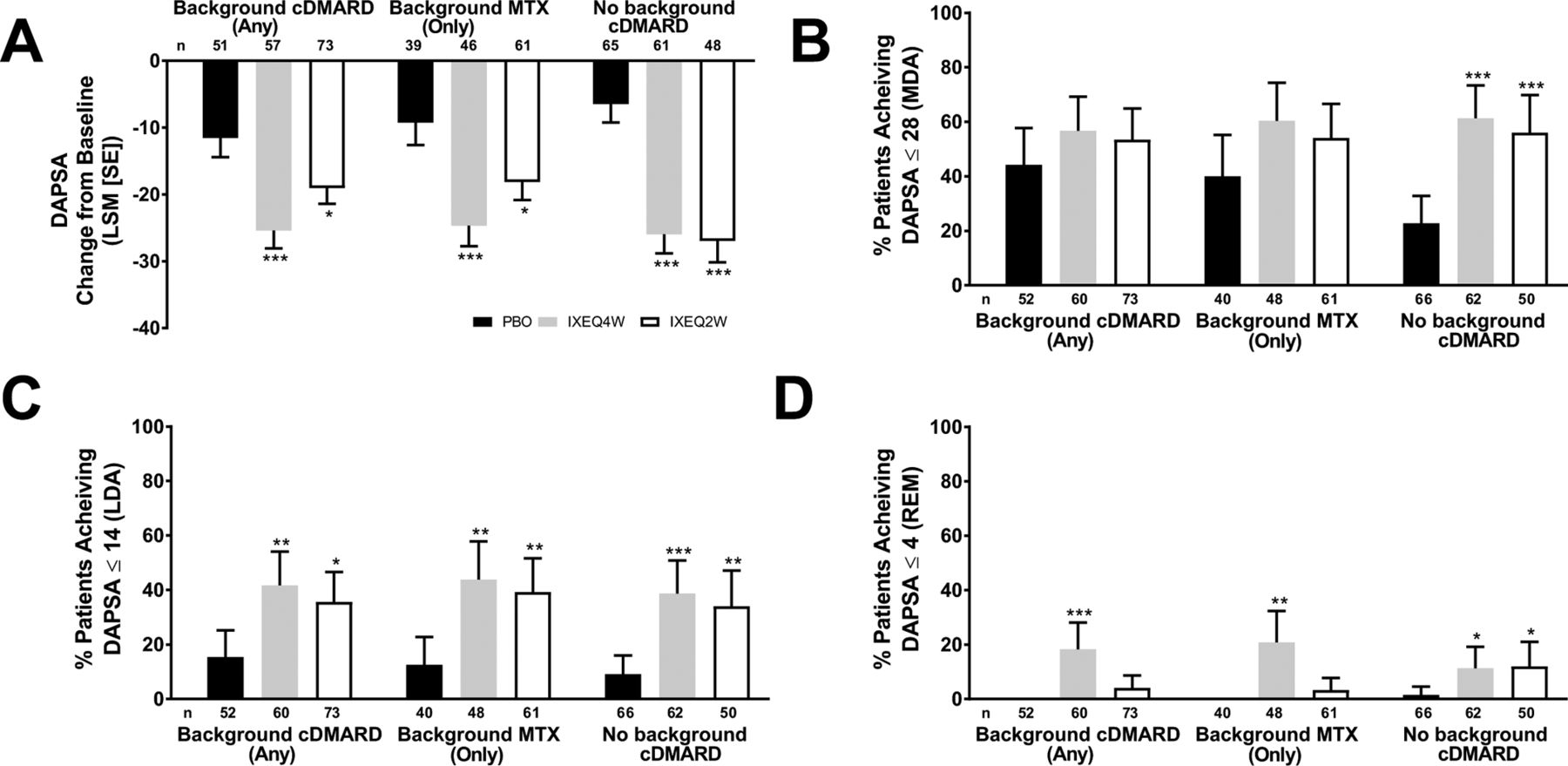
DAPSA LSM change from baseline (A), patients achieving DAPSA ≤28 (MDA) (B), DAPSA ≤14 (LDA) (C) or DAPSA ≤4 (REM) (D) after 24 weeks in patients treated with PBO, IXEQ4W or IXEQ2W alone or when added to background cDMARDs or MTX. DAPSA ≤28 (MDA) and DAPSA ≤14 (LDA) were calculated in a cumulative manner, thus if a patient was classified as achieving DAPSA ≤28 (MDA), the patient may also have achieved DAPSA ≤14 (LDA) and DAPSA ≤4 (REM). Percentages reported with 95% confidence intervals. The treatment-by-cDMARD use interaction p-value for DAPSA was 0.064, DAPSA ≤28 was 0.060, DAPSA ≤14 was 0.730, and DAPSA ≤4 was 0.049. *p<0.05, **p<0.01, ***p<0.001, all versus PBO. cDMARD, conventional disease-modifying antirheumatic drugs; DAPSA, Disease Activity Index for Psoriatic Arthritis; DAPSA ≤28 (MDA), DASPA moderate disease activity; DAPSA ≤14 (LDA), DAPSA low disease activity; DAPSA ≤4 (REM), DAPSA remission; IXEQ4W, ixekizumab every 4 weeks; IXEQ2W, ixekizumab every 2 weeks; LSM, least squares mean; MTX, methotrexate; n, number of patients; PBO, placebo.

DAS28-CRP LSM change from baseline after 24 weeks in patients treated with PBO, IXEQ4W or IXEQ2W alone or when added to cDMARDs or MTX. The treatment-by-cDMARD use interaction p-value for DAS28-CRP was 0.037. **p<0.01, ***p<0.001, both versus PBO. cDMARD, conventional disease-modifying antirheumatic drugs; DAS28-CRP, 28-joint Disease Activity Score using C-reactive protein; IXEQ4W, ixekizumab every 4 weeks; IXEQ2W, ixekizumab every 2 weeks; LSM, least squares mean; MTX, methotrexate; n, number of patients; PBO, placebo; SE, standard error.
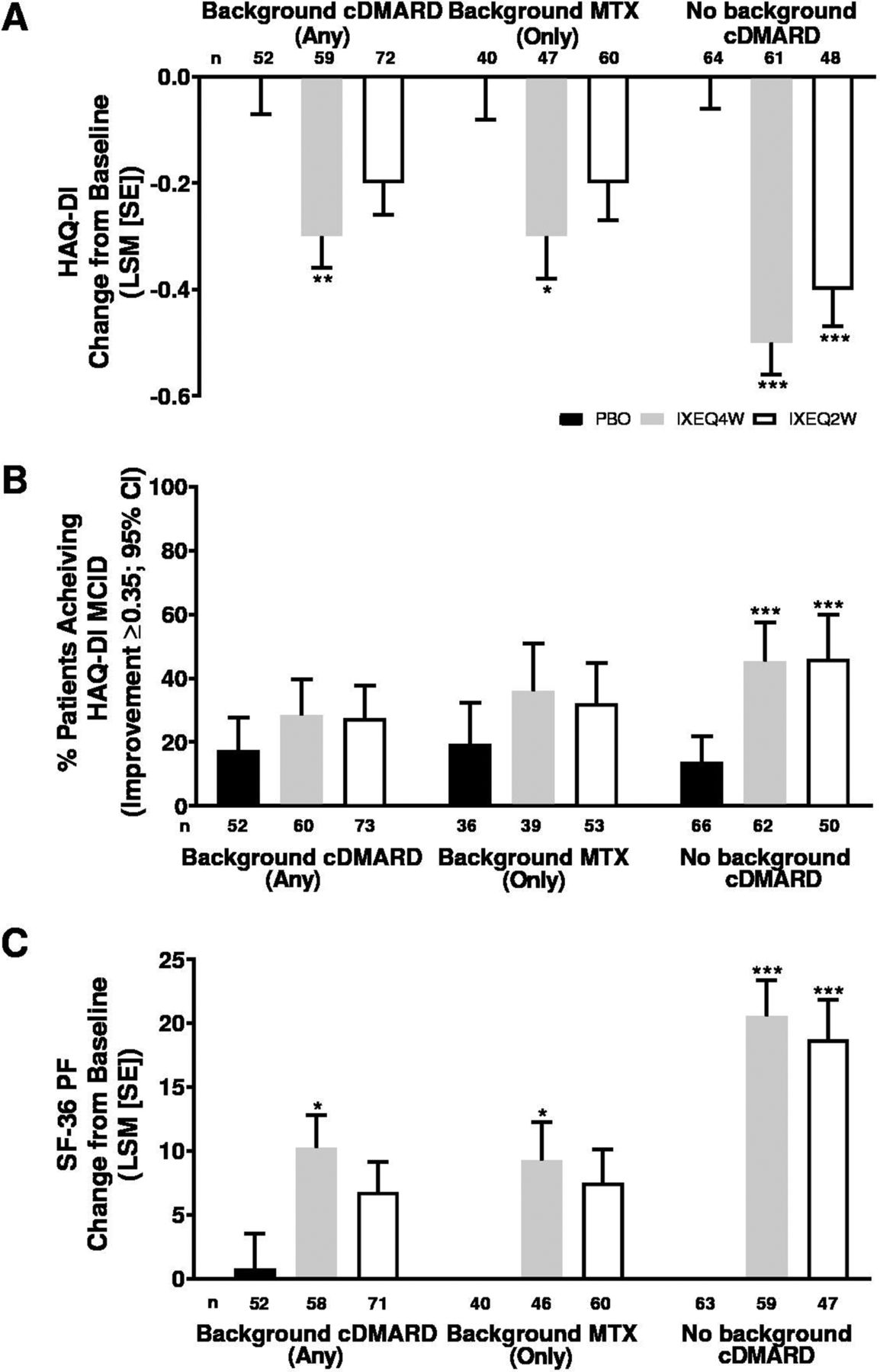
Physical function improved in ixekizumab-treated patients regardless of background cDMARD use at week 24 relative to PBO and reflected improvements in physical function observed in the overall trial population at week 24.13 IXEQ4W-treated patients achieved significant improvements in HAQ-DI change from baseline relative to PBO at week 24, whether they received ixekizumab alone or when added to background cDMARDs or MTX (figure 4A). IXEQ2W-treated patients also achieved significant improvements in HAQ-DI change from baseline relative to PBO at week 24 when they received IXEQ2W alone; the differences were not significant for the other two subgroups (when added to background cDMARDs or MTX), but ixekizumab showed numerically better improvements than PBO. The proportion of patients achieving HAQ-DI MCID was numerically higher relative to PBO at week 24, whether they received ixekizumab alone or when added to background cDMARDs or MTX, with significantly higher proportions observed in patients treated with IXEQ4W or IXEQ2W alone (figure 4B). Significant improvements were also observed in SF-36 PF change from baseline in patients treated with IXEQ4W, regardless of background cDMARD use (figure 4C). In patients treated with IXEQ2W, significant improvements in SF-36 PF change from baseline were observed in patients treated with monotherapy only.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
HAQ-DI LSM change from baseline (A), proportion of patients achieving HAQ-DI MCID (improvement ≥0.35) (B) and SF-36 PF LSM change from baseline (C) after 24 weeks in patients with PsA treated with PBO, IXEQ4W or IXEQ2W alone or when added to background cDMARDs or MTX. The treatment-by-cDMARD use interaction p-value was 0.040 for HAQ-DI, 0.1475 for HAQ-DI MCID, and 0.044 for SF-36 PF. *p<0.05, **p<0.01, ***p<0.001, all versus PBO. cDMARD, conventional disease-modifying antirheumatic drugs; CI, confidence interval; HAQ-DI, Health Assessment Questionnaire Disability Index; IXEQ4W, ixekizumab every 4 weeks; IXEQ2W, ixekizumab every 2 weeks; LSM, least squares mean; MCID, minimal clinically important difference; MTX, methotrexate; n, number of patients; PBO, placebo; SE, standard error; SF-36 PF, 36-item Short Form Survey physical functioning domain.
Safety was investigated across subgroups and it was consistent with the profile reported in the overall SPIRIT-P2 population,13 with a higher proportion of patients treated with IXEQ4W or IXEQ2W when added to background cDMARD or MTX reporting one or more TEAEs relative to PBO at week 24 (table 2). In the monotherapy subgroup, the frequency of TEAEs was higher with IXEQ2W versus PBO but comparable between IXEQ4W and PBO. SAEs were reported in up to 8.0% of patients across subgroups.
Safety overview of ixekizumab after 24 weeks of treatment when subdivided according to background cDMARD or MTX use at baseline
Serious infections were only reported in patients treated with IXEQ2W, but the frequencies were low (3.3% or less), and none were related to T uberculosis or C andida. Discontinuations due to AEs were reported in up to 10% of patients across subgroups.
Discussion
Ixekizumab was demonstrated to be efficacious relative to PBO at 24 weeks, whether used alone or when added to any background cDMARDs or only MTX in cDMARD-experienced patients with active PsA and previous inadequate response or intolerance to TNFis. In addition, the safety findings were consistent with previous safety reporting for ixekizumab in both psoriasis and PsA.12 13 20 21 Due to the study design, direct comparisons between subgroups were not possible. Notably, this study was the first pivotal PsA trial to exclusively enrol TNFi-experienced patients and it demonstrates that regardless of background cDMARD use, clinically meaningful efficacy is achieved in patients treated with ixekizumab relative to PBO.
The relative impact of concomitant cDMARDs on the overall biologic effectiveness in PsA is a controversial and discussed theme. Reports from previous RCTs are consistent with our results and demonstrate that with biologics in PsA, clinically meaningful efficacy is achieved with biologics alone or when added to background cDMARD or only MTX use. In SPIRIT-P1, biologic-naive patients treated with ixekizumab for 24 weeks achieved clinical efficacy relative to PBO regardless of background cDMARD use.9 Studies investigating etanercept6 or adalimumab5 treatment for 24 weeks reported efficacy in patients with PsA, regardless of background MTX status. None of these RCTs, including the ixekizumab study presented here, were designed to directly compare the efficacy of biologics alone to biologics combined with cDMARDs in cDMARD-naive patients, although such studies have been performed for rheumatoid arthritis as permitted by study design.22 Nonetheless, real-world data from observation studies and registries are a bit more controversial. In a long-term non-interventional observational study of patients with PsA, Behrens and colleagues observed that background MTX use did not impact efficacy outcomes.23 However, studies using the NOR-DMARD and DANBIO registries have reported similar efficacy findings, but have noted that drug survival is higher for some biologics (particularly for monoclonal antibodies such as infliximab) with background MTX use.7 24 These registry results indicate MTX may contribute to drug survival, perhaps by reducing immunogenicity as hypothesised by Fagerli and colleagues in combination with some biological therapies for PsA.7 24 Investigations into drug survival were not possible in this subset analysis due to the short-term study period, but would be informative in the context of a long-term RCT, observational or registry study.
Our study had limitations. Results represented here were derived from a subset analysis of SPIRIT-P2, and the majority were post hoc. Due to the study design and due to the fact that some patients were receiving cDMARDs at baseline, we were unable to make direct efficacy comparisons between ixekizumab treatment with and without cDMARD use. Furthermore, all enrolled patients were cDMARD-experienced per study inclusion criteria. Responses in cDMARD-naive patients may differ from those observed in this subset analysis.9 In addition, the size of the reported subgroups made it difficult to interpret the safety results, but we note they were consistent with the overall trial population. Nonetheless, the study results provide clinicians with additional information on the utility and safety profile associated with the use of ixekizumab either alone or when added to cDMARD use in an exclusively TNFi-experienced patient population with PsA.
Overall, we demonstrate that ixekizumab was efficacious, relative to PBO, when used alone or when added to cDMARDs in patients with active PsA and previous inadequate response or intolerance to TNFis. The frequencies of reported TEAEs, serious AEs (including serious infections) and discontinuations due to AEs were consistent with previous reports of ixekizumab use in patients with PsA and psoriasis.12 13 20 21
Acknowledgments
The authors acknowledge Brian S. Comer, PhD, an employee of Eli Lilly and Company, for medical writing support. The authors also thank Natacha Gallot, Justin Grondines and Elizabeth King, employees of ClinBAY, for statistical analysis and programming support.
References
Footnotes
Presented at The results of this study were previously presented, in part, at EULAR (European League Against Rheumatism) 2018.
Correction notice The article has been corrected since it first published online. The authors noticed some copyediting errors which have been rectified now.
Contributors PN, OB, DHA were involved in the conception and design of the clinical study; PN, A-MO, OB, DHA, AD were involved in the acquisition of the data. All authors were involved in the analysis and interpretation of the data. All authors were involved in the drafting and revision of the manuscript. SSR was involved in the statistical analyses.
Funding This study was funded and sponsored by Eli Lilly and Company.
Competing interests PN: Grant/research support from: AbbVie, Amgen, Bristol-Myers Squibb, Celgene, Eli Lilly and Company, Hospira, Janssen, MSD, Novartis, Pfizer, Roche, Sanofi, UCB; Consultant for: AbbVie, Amgen, Bristol-Myers Squibb, Celgene, Eli Lilly and Company, Hospira, Janssen, MSD, Novartis, Pfizer, Roche, Sanofi, UCB; Speakers bureau: AbbVie, Amgen, Bristol-Myers Squibb, Celgene, Eli Lilly and Company, Hospira, Janssen, MSD, Novartis, Pfizer, Roche, Sanofi, UCB. FB: Grant/research support from: Abbvie, Pfizer, Roche, Chugai, Prophylix, Novartis, Iron4U; Consultant for: Abbvie, Pfizer, Roche, Chugai, UCB, BMS, Celgene, MSD, Novartis, Biotest, Janssen, Genzyme, Sanofi, Eli Lilly and Company, Sandoz; Speakers bureau: Abbvie, Pfizer, Roche, Chugai, UCB, BMS, Celgene, MSD, Novartis, Biotest, Janssen, Genzyme, Sanofi, Eli Lilly and Company, Sandoz. A-MO: Grant/research support from: Abbvie, Celgene, Eli Lilly and Company, Horizon, Janssen, Novartis, Pfizer; Consultant for: Eli Lilly and Company, Janssen, Novartis, Pfizer, UCB. SSR, DHA, OB: All employees and shareholders of Eli Lilly and Company. AD: Grant/research support from: Pfizer; Consultant for: Pfizer.
Patient consent Patient consent was obtained.
Ethics approval The protocol was approved by each site’s institutional review board or ethics committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Eli Lilly provides access to relevant anonymised patient level data from studies on approved medicines and indications as defined by the sponsor-specific information on www.clinicalstudydatarequest.com. For details on submitting a request, see the instructions provided at www.clinicalstudydatarequest.com.