Article Text

Abstract
Objective To assess the long-term (3 year) efficacy and safety of secukinumab in patients with active psoriatic arthritis (PsA) in the extension phase of the FUTURE 1 study (NCT01892436).
Methods Following the 2-year core trial, eligible patients receiving subcutaneous secukinumab 150 or 75 mg entered a 3-year extension phase. Results are presented for key efficacy and safety endpoints at week 156.
Results In total, 460 patients entered the extension study; 308 patients originally randomised to secukinumab were assessed for efficacy. Sustained improvements in all efficacy endpoints were achieved with secukinumab through week 156. Overall, 76.8%/54.9% (secukinumab 150 mg) and 65.2%/39.0% (secukinumab 75 mg) of patients achieved an American College of Rheumatology (ACR) 20/50 response (multiple imputation data); ACR20 responses were sustained irrespective of previous anti-tumour necrosis factor exposure. Improvements in quality of life and physical function were also sustained through week 156. Radiographic results (observed data; van der Heijde modified total Sharp score (mTSS)) showed that 78.1% (secukinumab 150 mg) and 74.8% (secukinumab 75 mg) of patients had no radiographic progression (≤0.5 increase in mTSS) through week 156. Exposure-adjusted incidence rates for selected adverse events per 100 patient-years (secukinumab 150/75 mg) were serious infections (1.7/1.6), Candida infections (1.4/0.7), Crohn’s disease (0/0.3), ulcerative colitis (0/0.3) and major adverse cardiac events (0.3/0.8).
Conclusion Subcutaneous secukinumab provided sustained improvements in the signs and symptoms, quality of life and physical function of patients with active PsA with low rate of radiographic disease progression through 3 years. Secukinumab was well tolerated with no new safety signals.
- psoriatic arthritis
- secukinumab
- radiography
- IL-17A
- biological therapy
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Secukinumab, a fully human monoclonal IgG1κ antibody that selectively neutralises IL-17A, provided sustained improvements in the signs and symptoms of active psoriatic arthritis (PsA) in phase III FUTURE 1 study over 2 years.
What does this study add?
Secukinumab 150 mg (approved dose) sustained or further improved the clinical responses in patients with active PsA over 3 years in FUTURE 1 study, with a low rate of radiographic disease progression and safety profile consistent with previous reports.
How might this impact on clinical practice?
This long-term (3-year) data for secukinumab in PsA adds to the growing body of evidence supporting the use of IL-17 inhibitors in PsA, as recognised in the guidelines of European League Against Rheumatism and Group for Research and Assessment of Psoriasis and Psoriatic Arthritis recommendations.
Introduction
Psoriatic arthritis (PsA) is a chronic, inflammatory disease characterised by peripheral arthritis, axial disease, dactylitis, enthesitis, and skin and nail psoriasis.1 It can be associated with more serious comorbidities such as cardiovascular disease, metabolic syndrome and diabetes,2–4 and has a substantial impact on quality of life, physical function and work productivity.5 Management of PsA should ideally aim to minimise articular pain, enthesitis, structural damage, disability and dermatological symptoms.1 6–8 The introduction of anti-tumour necrosis factor-α inhibitors (anti-TNF agents) has significantly improved outcomes among patients with PsA,6 9–11 but a proportion of patients have an inadequate response or poor tolerability to these agents.3 12 Secukinumab, a fully human monoclonal antibody that selectively neutralises interleukin IL-17A,13 has been shown to have significant efficacy in the treatment of ankylosing spondylitis,13 14 moderate to severe psoriasis15 and PsA,16–18 demonstrating a rapid onset of action and sustained responses with an acceptable safety profile across the three indications.19 20
In the randomised, placebo-controlled, phase III FUTURE 1 core study, secukinumab demonstrated rapid, significant and sustained reductions in the signs and symptoms of PsA, and inhibited radiographic progression through 2 years of treatment.16 21 The ongoing FUTURE 1 extension trial is a 3-year extension of the 2-year core trial, assessing the effect of secukinumab on the signs and symptoms, structural damage, physical function and quality of life of patients with active PsA, in addition to evaluating long-term safety. Here, we present efficacy and safety results of all doses (secukinumab 150 mg (approved dose) and 75 mg) assessed in the FUTURE 1 study through 3 years (2 years of the core study plus 1 year of the extension).
Methods
Study population
Detailed patient eligibility criteria have been reported previously.16 Briefly, patients were ≥18 years, with active PsA according to the Classification Criteria for Psoriatic Arthritis,22 with moderate to severe symptoms for ≥6 months, ≥3 tender joints of 78 and ≥3 swollen joints of 76, despite previous treatment with nonsteroidal anti-inflammatory drugs, disease-modifying anti-rheumatic drugs or anti-TNF agents. Concomitant use of oral glucocorticoids (up to 10 mg/day of prednisone or equivalent) and methotrexate (up to 25 mg/week) was permitted. Patients were excluded if they had previous therapy with biologic agents other than anti-TNF agents, treatment with more than three anti-TNF agents, active inflammatory disease other than PsA or active/history of ongoing infection. The study was approved by the institutional review board or ethics committee at each participating site (Swedish Medical Centre and University of Washington (1143210), Dallas VAMC and University of Texas Southwestern Medical Centre (#IRB 14-025), Barts Health NHS Trust (13/WM/0365), University of Erlangen-Nuremberg (AZ: EK-13/081), Monash University (2013-08-423), University of Hasselt and Maastricht University Hospital (2013/1062); online supplementary table 1) and was conducted according to the Declaration of Helsinki.23
Supplemental material
Study oversight and design
The randomised, double-blind FUTURE 1 study (NCT01892436) was conducted across 104 sites in North and South America, Europe, the Middle East, Australia and Asia, the design of which has been previously reported.16 Briefly, eligible patients were initially randomised to receive secukinumab 10 mg/kg intravenous (IV) loading dose at baseline and weeks 2 and 4, followed by subcutaneous (SC) secukinumab 150 mg or 75 mg every 4 weeks from week 8. Placebo was given using the same IV-to-SC dosing schedule. At week 16, placebo-treated patients were re-randomised to receive SC secukinumab 150 mg or 75 mg from either week 16 for placebo non-responders (<20% improvement from baseline in both tender and swollen joints) or week 24 for placebo responders. Randomisation was stratified by previous anti-TNF status, with patients being anti-TNF-naïve or anti-TNF-experienced (incomplete responders who had active disease despite having received an anti-TNF agent for ≥3 months, or who had stopped treatment due to safety and tolerability reasons). At the end of the 2-year core trial, eligible patients entered the 3-year extension phase. From week 156, patients not adequately responding to secukinumab 75 mg could be up-titrated to secukinumab 150 mg, at the discretion of investigators. See online supplementary figure 1 for the extension study design.
Supplemental material
Endpoints and assessments
Efficacy results at week 156 are reported for patients who were originally randomised to 10 mg/kg IV loading followed by SC 150 mg and 75 mg maintenance with secukinumab. The primary efficacy endpoints of this extension study were the proportion of patients treated with secukinumab 150 mg and 75 mg achieving an American College of Rheumatology (ACR) 20/50/70 response over time.24 Week 156 assessments also included the proportion of patients achieving Psoriatic Area and Severity Index (PASI) 75 responses, resolution of dactylitis and enthesitis and proportion of patients with minimal disease activity (MDA). Changes from baseline in Disease Activity Score in 28 joints using C reactive protein (DAS28-CRP), Health Assessment Questionnaire–Disability Index (HAQ-DI), Medical Outcome Short Form (36) Health Survey (SF-36) Physical Component Summary (PCS) and Mental Component Summary (MCS), and radiographic progression (measured by the van der Heijde modified total Sharp score (mTSS), which is the sum of erosion and joint space narrowing (JSN) scores).25 X-rays of the hands, wrists and feet, performed at baseline and week 156, were read centrally by two independent readers blinded to the treatment arms. The mean score was used for all analyses. Radiographic non-progressors were defined as patients with a change from baseline of ≤0.5 in mTSS during the considered period, as per guideline recommendations.26 Long-term safety and tolerability was assessed over the treatment period by monitoring the frequency of treatment-emergent adverse events (AEs), serious AEs, laboratory abnormalities and vital signs.
Statistical analyses
Sample size calculation estimated that 600 patients (200 in each secukinumab group and 200 in the placebo group) were sufficient to meet primary and key secondary endpoints. Analysis of primary and other efficacy endpoints of the core trial have been reported previously.16 It was estimated that 460–520 patients would be eligible for enrolment in this extension study.
Evaluations of efficacy were performed on patients enrolled in the extension with at least one efficacy assessment during the extension and who were originally randomised to 10 mg/kg IV loading at baseline and weeks 2 and 4, followed by SC 150 mg and 75 mg maintenance with secukinumab starting at week 8. Efficacy data from patients who discontinued the study were considered as end of treatment period results. No formal hypotheses were planned in this extension study. In the current analysis, missing binary variables up to week 156 were imputed using multiple imputation. Summaries of continuous variables are as observed and presented as mean±SD. Dactylitis and enthesitis were evaluated using multiple imputation in the subgroup of patients with these symptoms at baseline. PASI response was evaluated in the subgroup of patients with at least 3% of the body surface area affected by psoriatic skin involvement.
Joint structural damage (changes from baseline in mTSS scores, erosion scores and JSN scores) and proportion of radiographic non-progressors were analysed using observed data based on all evaluable patients with data at both baseline and week 156.
Predefined subgroup analyses were carried out on the basis of previous anti-TNF therapy (anti-TNF-naïve or anti-TNF-experienced) for key efficacy endpoints.
Safety analysis included all patients (core and extension study phase) who received ≥1 dose of secukinumab, with patients analysed for safety according to the actual treatment they received. As safety was collected over the treatment period and included data past week 156, safety analyses included patients who up-titrated from 75 mg to 150 mg; therefore, patients who were up-titrated were counted in either secukinumab group depending on the timing of the event. Data for selected AEs are presented as ted incidence rates (EAIRs) exposure-adjusted incidence rate (EAIR) per 100 patient-years.
Results
Patients
Of the 606 patients originally randomised in the FUTURE 1 core trial, 460 (75.9%) patients entered the extension study (secukinumab 150 mg (N=162), secukinumab 75 mg (N=148), placebo then secukinumab (N=150)). Figure 1 shows a breakdown of patients who entered the extension study. Three patients who were originally randomised to secukinumab and who discontinued the study were not part of the efficacy analyses due to the lack of efficacy assessment during the extension study; reasons for discontinuation were sudden cardiac death (150 mg arm), patient decision (75 mg arm) and development of squamous cell carcinoma of the pharynx (75 mg arm).

Patient disposition up to week 156. FAS, full analysis set; n, number of patients; SC, subcutaneous.
Within the full analysis set (FAS) population, the reasons for discontinuation included patient decision (n=7), AEs (n=5), lack of efficacy (n=5), physician decision (n=2), pregnancy (n=2) and loss to follow-up (n=1). The patient demographics and baseline disease characteristics of patients entering the long-term extension phase were similar across the 150 mg and 75 mg arms (table 1). For secukinumab 150 mg and 75 mg, more than half the patients (55.3% and 55.8%) had psoriasis affecting at least 3% of their body surface area; 51.6% and 52.4% had dactylitis and 61.5% and 61.9% had enthesitis, respectively. A total of 25.5% and 25.2% were anti-TNF-experienced; 18.6% and 16.3% had received one anti-TNF therapy and 6.8% and 8.8% had received two or more anti-TNF therapies in the secukinumab 150 mg and 75 mg arms, respectively. A total of 60.9% and 61.9% were receiving concomitant methotrexate in the secukinumab 150 mg and 75 mg arms, respectively.
Demographics and baseline characteristics of patients originally randomised to secukinumab who entered the extension phase
Efficacy
Out of 460 (75.9%) patients who entered the extension trial, 435 (95.2%) completed 156 weeks (ie, 52 weeks of the extension). Sustained improvements in efficacy were observed during this extension study, with 76.8% and 65.2% of patients treated with secukinumab 150 mg and 75 mg achieving an ACR20 response and 54.9/32.9% and 39.0/26.0% achieving ACR50/70 responses through week 156, respectively (figure 2A and table 2). ACR20 response rates were sustained through week 156, irrespective of previous anti-TNF agent exposure, with ACR20 responses achieved by 81.0% and 67.3% of anti-TNF-naïve patients and 61.5% and 55.6% of anti-TNF-experienced patients treated with secukinumab 150 mg and 75 mg, respectively (figure 2B). Resolution of dactylitis and enthesitis were sustained through week 156, with 88.1% and 86.8% of patients achieving dactylitis resolution and 76.7% and 74.8% achieving enthesitis resolution in the 150 mg and 75 mg arms, respectively (table 2 and online supplementary figure 2). Secukinumab also demonstrated sustained improvement in PASI 75 responses and DAS28-CRP through week 156 (table 2). Proportion of patients with MDA response at week 156 was 43% and 33% in secukinumab 150 mg and 75 mg, respectively (table 2). Improvements in physical and mental component of quality of life (SF-36 PCS and SF-36 MCS) and in physical function (HAQ-DI) were sustained over the treatment period for secukinumab 150 mg and 75 mg (table 2).
Supplemental material

{kind=link}
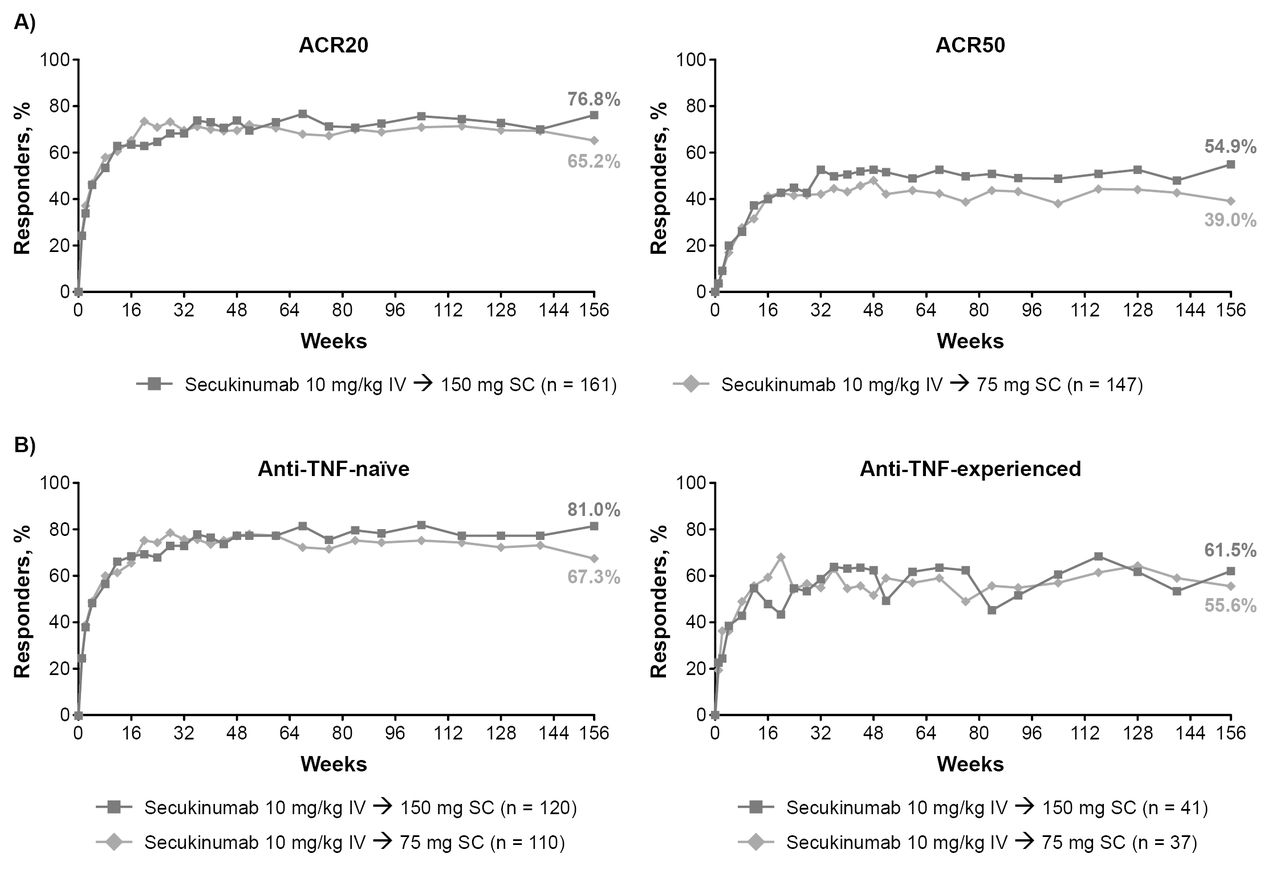{kind=link}
(A) ACR20/ACR50 response rates through week 156 in all patients originally randomised to secukinumab. (B) ACR20 response rates through week 156 in anti-TNF-nave and anti-TNF-experienced patients. Multiple imputation applied to missing variables through week 156. ACR, American College of Rheumatology; IV, intravenous; n, number of patients; SC, subcutaneous; TNF, tumour necrosis factor.
Secondary efficacy endpoints at weeks 52, 104 and 156 in patients originally randomised to secukinumab
Results from patients with evaluable X-ray assessments demonstrated that 78.1% (100/128) and 74.8% (92/123) of patients had no radiographic progression (mTSS score ≤0.5) from baseline to week 156, in the 150 mg and in the 75 mg arms, respectively (table 3A). Mean changes in mTSS score from baseline to week 156 for patients in the 150 mg and 75 mg arms are shown in table 3B. Subgroup analysis by anti-TNF status demonstrated that in patients with evaluable X-ray assessments, 78/100 (78.0%) and 73/94 (77.7%) of anti-TNF-naïve patients and 22/28 (78.6%) and 19/29 (65.5%) of anti- TNF-experienced patients were radiographic non-progressors in the 150 mg and 75 mg groups, respectively (table 3A). Changes in mTSS score from baseline to week 156 in anti-TNF-naïve patients remained low (table 3B).
Proportion of radiographic non-progressors
Mean change in X-ray assessments from baseline to week 156 (mTSS score, erosion score and JSN score—evaluable cases, observed data)
Safety
The incidence of AEs and serious AEs are presented in table 4. The EAIR per 100 patient-years of AEs in the any secukinumab 150 mg and 75 mg arms was 158.8 and 128.9; the rate of serious AEs was 9.3 and 6.4, respectively. The most commonly reported AEs with secukinumab were infections and infestations, musculoskeletal and connective tissue disorders and gastrointestinal disorders, which were stable over the long-term period of this study. EAIRs for serious infections and infestations, Crohn’s disease, ulcerative colitis, major adverse cardiovascular events (MACE), inflammatory bowel disease and neoplasms (benign, malignant or unspecified) for secukinumab 150 mg and 75 mg were consistent with previous trials and are reported in table 4. Candida infections were reported in 12 (EAIR=1.4) and 5 (EAIR=0.7) patients in the any secukinumab 150 mg and 75 mg arms, respectively. All cases of Candida infection were mild to moderate in severity and resolved spontaneously with standard treatment or with non-drug therapy; no cases led to discontinuation of secukinumab treatment. One case of reactivation of latent tuberculosis was reported in the any secukinumab 150 mg arm.
AEs and serious AEs across entire treatment period
Deaths (spontaneous acute myocardial infarction (n=1) and cardiac failure (n=1)) were reported in two patients with concomitant disease in the any secukinumab 150 mg arm. Deaths occurred in three patients in the any secukinumab 75 mg arm including two patients (stroke (n=1) and myocardial infarction (n=1)) with concomitant disease and in one patient who developed squamous cell carcinoma during the study and discontinued secukinumab treatment on receiving the diagnosis.
Discussion
Results from the first year of the phase III FUTURE 1 extension study have furthered our understanding of the role of secukinumab in the long-term treatment of PsA. Secukinumab demonstrated sustained improvements in the signs and symptoms, function and health-related quality of life in patients with active PsA, in addition to low rate of radiographic disease progression through 3 years of treatment. Patient retention rates throughout this trial were high with 95.2% of the FAS population completing the 156-week treatment period, supporting the sustained efficacy and tolerability of secukinumab.
These are the first 3-year findings reported for secukinumab in PsA and the data add to the growing body of evidence supporting the use of IL-17 inhibitors in PsA, as recognised in the guidelines from the European League against Rheumatism and Group for Research and Assessment of Psoriasis and Psoriatic Arthritis management recommendations.7 27
Consistent with previous reports, clinical benefits with secukinumab were observed regardless of prior exposure to anti-TNF therapy,28 with better responses observed in anti-TNF-naïve patients supporting it use as a treatment for patients with PsA naïve to anti-TNF therapy and those who have experienced an inadequate response or intolerance to these agents.6 29–32
The IL-17 pathway plays a role in irreversible structural damage experienced during inflammatory arthritis,33–36 as indicated by the increased presence of IL-17+CD8+ T-cells in the joints of patients with PsA.33 Previously published results from the FUTURE 1 study have demonstrated that secukinumab significantly inhibited radiographic progression in patients with PsA versus placebo at week 24,37 with sustained inhibition through 2 years.16 28 Results reported here extend these findings up to 3 years of treatment.
The safety profile of secukinumab in this trial was consistent with previous reports in patients with PsA and moderate-to-severe plaque psoriasis, with no new or unexpected safety signals observed.15–17 The most common AEs observed (infections and infestations, musculoskeletal and connective tissue disorders, and gastrointestinal disorders) and serious AEs are consistent with data from previous reports in patients receiving secukinumab and placebo.16
Patients with PsA have been reported to have a possible elevated risk of cardiovascular disease38–40 when treated with anti-IL-12/23 agents,41 42 and thus cardiovascular AEs are of interest in patients treated with biological agents. In our study, the EAIRs of MACE were 0.3 and 0.8 in the any secukinumab 150 mg and 75 mg groups, respectively. This is in line with previously reported rates of MACE in secukinumab-treated patients, including those from the FUTURE 1 core study,15 16 28 and corroborate results from a recent meta-analysis showing a low overall MACE rate in patients with psoriasis who were treated with the IL-17A inhibitors, secukinumab and ixekizumab.43 Candida infections observed during this study are consistent with results from phase III studies, including the FUTURE 1 core study, in patients with PsA17 28 and psoriasis.15 All cases were mild to moderate in severity, did not lead to discontinuation and were managed with standard antifungal therapy. Increased rate of Candida infections is likely attributable to the role of IL-17 in host defense against fungal infections, particularly at mucosal sites.44 Other AEs of special interest in this study, including Crohn’s disease and ulcerative colitis, were low and in line with previously reported findings, including those from the FUTURE 1 core study.16 20 They also confirm results from a recent review of phase II and III studies where patients with PsA treated with secukinumab (up to 112 weeks) had EAIRs of 0.07 for Crohn’s disease and 0.14 for ulcerative colitis.45 No suicides or suicide ideation were reported during this study.
While this study was not specifically designed to examine the psychological impact of secukinumab treatment on patients with PsA, an improvement in emotional role functioning and mental health was observed with secukinumab treatment, as assessed by the patient-reported SF-36 MCS.46
Limitations of this study included a lack of a long-term comparator due to the fact that long-term treatment with placebo is considered unethical; the placebo-controlled period of the core trial was, therefore, only up to week 16. There was no active comparator included and results could be potentially biased as patients remaining in the study are those patients benefiting from secukinumab. While efficacy responses in the current study were sustained irrespective of previous anti-TNF exposure, patients eligible for inclusion in this study could have been treated with no more than three anti-TNF agents; this may be viewed as a limitation of the study.
In summary, secukinumab provided sustained improvements across multiple domains of PsA including signs and symptoms, quality of life and physical function in patients with active disease, in addition to low rate of radiographic disease progression through 3 years of treatment. The results confirmed a favourable and consistent safety profile through 3 years, which is comparable with that reported through 2 years in the core trial. Overall, the results of this extension study provide further supporting evidence for the long-term use of secukinumab in the treatment of patients with PsA.
Acknowledgments
This analysis was supported by Novartis Pharmaceuticals Corporation. The authors thank Gillian Brodie, Novartis Ireland Ltd, Ireland and Niladri Maity, Novartis, India for providing medical writing support, which was funded by Novartis in accordance with Good Publication Practice (GPP3) guidelines.
References
Footnotes
Contributors All authors participated in the interpretation of data, critical review and final approval of the manuscript.
Funding The study was sponsored by Novartis Pharma AG, Basel, Switzerland, and designed by the scientific steering committee and Novartis personnel. Medical writing support was funded by Novartis.
Competing interests PJM has received research grants from AbbVie, BMS, Celgene, Janssen, Lilly, Novartis, Pfizer, SUN, and UCB; consulting fees from AbbVie, Amgen, BMS, Celgene, Covagen, Crescendo , Janssen, LEO, Lilly, Merck, Novartis, Pfizer, SUN and UCB; and speakers’ bureau fees for AbbVie, Amgen, BMS, Celgene, Genentech, Janssen, Lilly, Pfizer and UCB. AK served as consultant for Novartis. AR has received research grants from Janssen, Novartis, Pfizer and AbbVie; and consulting fees from Lilly. HT served as consultant or participation in advisory boards for AbbVie, Novartis, Pfizer, UCB, Eli-Lilly, Janssen Education Grants, Novartis and Pfizer. JR received speaker fees from AbbVie, AstraZeneca, Biogen, BMS, Celgene, Chugai, GSK, Janssen, MSD, Novartis, Pfizer, Roche, Sanofi Aventis and UCB; consulting fees from AbbVie, AstraZeneca, Biogen, BMS, Chugai, MSD, Novartis, Pfizer, Roche, Sanofi Aventis and UCB. PG participated in clinical studies, advisory boards, received speaker’s fees from Abbott, Amgen, BMS, Lilly, MSD, Novartis, Pfizer, Roche, UCB and Will-Pharma. PP is an employee of Novartis with Novartis stock. EMD is an employee of Novartis. SM is an employee of Novartis, with Novartis stock. LP is an employee of Novartis with Novartis stock.
Patient consent Obtained.
Ethics approval This study was approved by the institutional review board or ethics committee at each participating site (Swedish Medical Centre and University of Washington (1143210), Dallas VAMC and University of Texas Southwestern Medical Center (IRB no. 14-025), Barts Health NHS Trust (13/WM/0365), University of Erlangen-Nuremberg (AZ: EK-13/081), Monash University (2013-08-423), University of Hasselt and Maastricht University Hospital (2013/1062); online supplementary table 1) and was conducted according to the Declaration of Helsinki.
Provenance and peer review Not commissioned; externally peer reviewed.
Data statement Novartis is committed to sharing with qualified external researchers, access to patient-level data and supporting clinical documents from eligible studies. These requests are reviewed and approved by an independent review panel on the basis of scientific merit. All data provided is anonymized to respect the privacy of patients who have participated in the trial in line with applicable laws and regulations.