Article Text

Abstract
The reading of acetylation marks on histones by bromodomain (BRD) proteins is a key event in transcriptional activation. Small molecule inhibitors targeting bromodomain and extra-terminal (BET) proteins compete for binding to acetylated histones. They have strong anti-inflammatory properties and exhibit encouraging effects in different cell types in vitro and in animal models resembling rheumatic diseases in vivo. Furthermore, recent studies that focus on BRD proteins beyond BET family members are discussed.
- rheumatoid arthritis
- inflammation
- fibroblasts
- autoimmune diseases
- systemic sclerosis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0
Statistics from Altmetric.com
Key messages
Inhibition of bromodomain and extra-terminal (BET) proteins (BRD2, BRD3, BRD4) has anti-inflammatory effects.
BET protein inhibition alleviates arthritis and fibrosis.
Bromodomain proteins of distinct families are important in Th17 cell differentiation.
There is a need for studies beyond the use of pan-inhibitors to unravel the role of individual bromodomain proteins.
Introduction
Acetylation of histones is associated with the establishment of euchromatin, which is a prerequisite for the activation of gene transcription and expression. The opposing functions of histone acetyl transferases (HAT) and histone deacetylases (HDAC) control the acetylation of lysine residues on histones but both protein families have also been shown to act on non-histone proteins.1 HDAC inhibitors have been widely studied in rheumatic diseases (RMD) in vitro and in vivo, underscoring the importance of protein lysine acetylation in this context.2 3
The recognition of ε-N-acetylation modifications on histones is a key event in the reading of epigenetic marks. Bromodomains (BRD) exclusively recognise acetylation motifs. BRDs form a bundle of four alpha helices, linked by interhelical loop regions of variable length and sequence that form the hydrophobic lysine acetlyation binding site and contribute to substrate specificity. The structural features of BRDs are in sharp contrast to other epigenetic reader domains such as chromodomains which hold a β-barrel structure.4 Many BRDs have good druggability, based on prediction from available structures and computational analysis.5 Frequently, BRDs are flanked by several other epigenetic reader domains, in multidomain BRD proteins, or they occur in tandem arrangement in bromodomain and extra-terminal domain (BET) proteins (BRD2, BRD3, BRD4, BRDT).6
Reading of acetylation marks by BRD enables the regulation of gene expression through a wide range of activities. Either BRD proteins act as scaffolds that enable the recruitment and binding of large protein complexes or they function as transcription factors and coregulators themselves. Additionally, BRD proteins can contain several catalytic domains enabling them to act as methyltransferases, ATP-depending chromatin remodelling complexes or HATs and helicases.7
Involvement of BRD proteins in diseases
The human genome encodes 61 BRDs that are present in 46 diverse proteins. These proteins cluster into eight major BRD families (I–VIII),6 with BET proteins being the foremost studied group. Dysfunction of BRD proteins has been linked to the development of several diseases. Mutations in BRD proteins, copy number alterations, misexpression and BRD-containing fusion proteins, resulting from chromosomal translocation, were observed in several cancers.7 Targeting BRD proteins has transitioned to the centre of epigenetic drug development in the last decade. Even some of the beneficial effects of HDAC inhibitors might be attributed to BRD, since HDAC inhibitors can induce a selective depletion of BRD proteins from the cells’ nuclei.8
BET protein inhibitors
In 2010, the first small molecule inhibitors targeting BET proteins, JQ1 and I-BET, have been described by Filippakopoulos et al and Nicodeme et al, which exhibited anticancer and anti-inflammatory activities, respectively.9 10 The availability of inhibitors has led to a significant increase in the BRD protein research. BET inhibitors (figure 1), targeting BRD2, BRD3, BRD4, and the testis-specific BRDT have been tested in many cell types, diseases and disease models suggesting a role of BET proteins far beyond cancer, such as inflammation,10–18 fibrosis,19–23 asthma 24 25 , coronary artery26 and cardiovascular disease,27 28 Alzheimer’s disease,29 30 autism-like syndrome,31 graft-versus-host disease32 and a variety of autoimmune conditions.33–37 BET inhibitors bind to the proteins’ BRD, compete for binding to acetylated histones and displace BET proteins from nuclear chromatin. This prevents the transcription of BRD protein target genes (figure 2). By now, at least 14 different small molecule inhibitors targeting BRD proteins have entered clinical trials for different forms of cancer, but also for the treatment of type 2 diabetes mellitus, cardiovascular disease and coronary artery disease.7 10

Inhibitors and their chemical structures targeting BET proteins. BD2, bromodomain 2; BET, bromodomain and extra-terminal.

Inhibitors, such as JQ1 and I-BET, targeting BET proteins compete with the BRD for binding to acetylated histones. Thereby, they prevent the transcription of their target genes depending on the cell type. This leads to decreased proliferation, suppressed cell differentiation and maturation and altered gene expression. BET inhibitors lead to decreased inflammation, cartilage destruction, bone resorption and fibrosis. AC, acetylated; BET, bromodomain and extra-terminal; BRD, bromodomain; col, collagen; MMP, matrix metalloproteinase; αSMA, alpha smooth muscle cell actin.
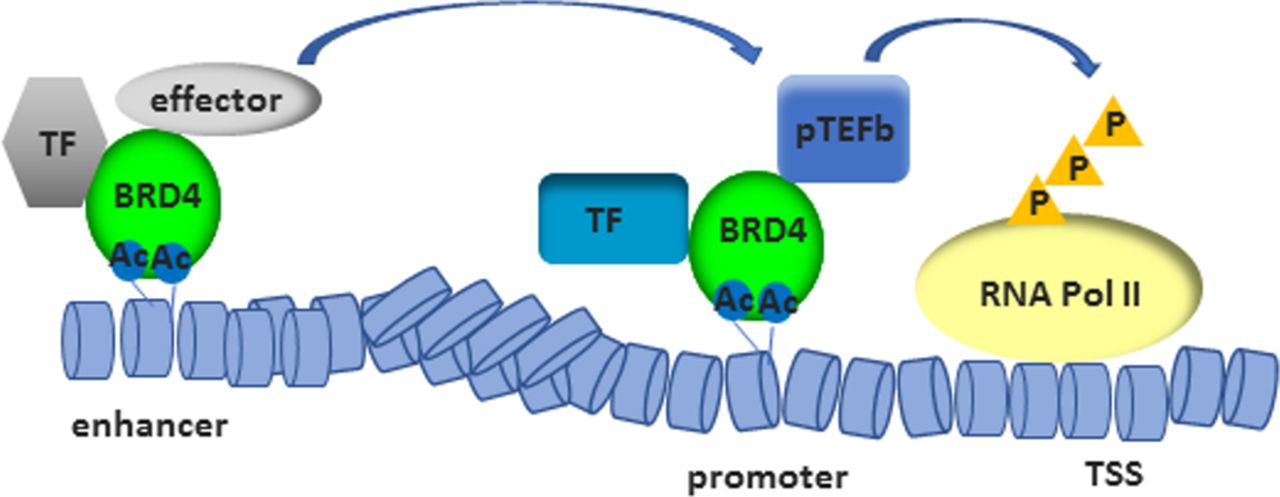
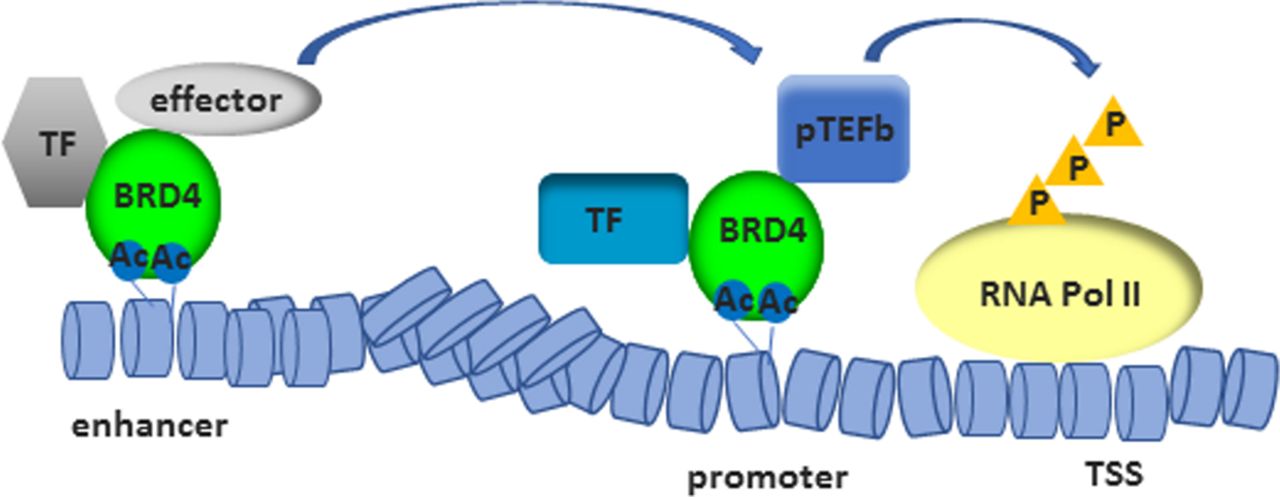
The most-studied member among BET proteins is BRD4. BRD4 confers the ability to direct the assembly of HAT-dependent chromatin complexes (figure 3), leading to the recruitment of positive transcription elongation factor b (p-TEFb)38 39 and subsequent phosphorylation of RNA polymerase II (RNA Pol II).40 Additionally, p-TEFb-independent transcriptional activation via the extra-terminal domain of BRD4 has been shown.41 BRD4 has also kinase activity and binds and phosphorylates RNA Pol II42 and acetylates H3K122, a residual critical for nucleosome stability, whose acetylation leads to histone eviction.43 Furthermore, by binding BRD4 to the acetylated lysine-310 of p65, BRD4 regulates the transcriptional activity of nuclear factor kappa light chain enhancer of activated B cells (NF-κB). This interaction was shown to be sensitive to treatment with JQ1, which induced the ubiquitination and subsequent degradation of the constitutively active nuclear form of p65.44 This data suggests an active role of BRD4 in transcription. Recent work has demonstrated the recruitment of BRD4 to enhancers45 and its involvement in assisting transcriptional elongation at enhancers (enhancer RNA (eRNA)) and gene bodies.46 Therefore, BET inhibitors have the potential for a dual targeting of gene expression in RMD, by affecting the inflammatory environment and by altering cell-type specific enhancer activity.9 47 Treatment with JQ1 was shown to preferentially affect the expression of super enhancer-associated gene transcripts compared with genes associated with bound BRD4 to normal enhancers.47 Super enhancers are cell type specific and consist of clusters of enhancers that span a long range of DNA (>10 kb). They are densely occupied by master transcription factors and histone marks and often guide eRNA expression.48 49 Super enhancers were demonstrated to specifically regulate genes associated with cell identity and disease.47 Tumour necrosis factor alpha (TNFα) was shown to be sufficient to alter the enhancer landscape of vascular endothelial cells, with a BRD4 eviction from basal super enhancers and a redistribution to de novo enhancers as well as super enhancers in a p65-dependent manner.50 Also, I-BET was reported to primarily affect the expression of genes from de novo enhancers, whereas promoters enriched for histone 3-acetylation and histone 4-acetylation already at basal levels (primed loci) were less affected.10 Likewise, a disease-specific, inflammation-associated super enhancer signature was identified in synovial fluid-derived CD4+ memory/effector T cells from patients with juvenile idiopathic arthritis (JIA). JQ1 treatment of these cells inhibited immune-related super enhancer activity and preferentially suppressed disease-associated gene expression. In contrast, the enhancer signature of peripheral blood-derived CD4+ memory/effector T cells from patients with JIA was relatively similar to those from healthy controls and less affected by JQ1 treatment.51 It remains to be elucidated whether the altered enhancer profile is causally related to disease pathogenesis or the consequence of the exposure to a chronic inflammatory milieu.
{kind=link}
{kind=link}
{kind=link}
By binding to acetylated histones, BRD4 contributes to the assembly of histone acetyltransferase-dependent chromatin complexes at gene promoters and to the recruitment of TF and p-TEFb. This leads to the phosphorylation of RNA Pol II. BRD4 binds also acetylated histones in enhancers and assists transcriptional elongation at enhancers and gene bodies. AC, acetylated; BRD4, bromodomain protein 4; P, phosphorylation; p-TEFb, positive transcription elongation factor B; RNA Pol II, RNA polymerase II; TF, transcription factors; TSS, transcriptional start site.
BET inhibitors can target a variety of cell types in RMD and have been tested in different mouse models resembling different aspects of RMD (table 1). Several different compounds are used in RMD research. Inhibitors in this review are named based on their description in the respective original paper. BET inhibitors block the maturation and differentiation of several cell types, including T cell subsets,33 34 52 53 dendritic cells54 and osteoclasts.55 56 BRD protein members of three distinct families have been shown to interfere with the differentiation and function of Th17 cells,34 37 52 57 58 suggesting a broad role of BRD proteins in autoimmune disorders. Furthermore, BET inhibitors affect the inflammatory responses of B cells,59 macrophages,10 11 13 synovial fibroblasts (SF)15 60 61 and chondrocytes.62 63
Inhibitors targeting BET proteins have been tested in animal models resembling diverse characteristic of rheumatic diseases
BET protein inhibition in macrophages
Treatment of murine bone marrow-derived macrophages (BMDM) with I-BET or JQ1 suppressed the expression of key lipopolysaccharide (LPS)-inducible cytokines, chemokines and transcription factors.10 11 13 In line with the in vitro data, injection of I-BET or JQ1 before the initiation of LPS-induced or Salmonella typhimurium-induced endotoxic shock was able to prevent or attenuate death of mice.10 11 Anti-inflammatory effects of BET inhibitors were later also confirmed in human monocytes,36 64 where I-BET151 diminished both autocrine interferon (IFN)-β production and transcriptional responses to IFN-β.64 Some of these effects might be attributed to the function of BRD3, since the knockout of BRD3 in murine RAW246.7 macrophages inhibited the virus-triggered IFN-β production.65
Whereas whole transcriptome data is available of LPS-stimulated BMDM treated with I-BET10 or JQ1,13 respectively, very little is known on the individual roles of BRD2, BRD3 and BRD4 on regulating the inflammatory response of BMDM and only few direct target genes are known. Individual silencing of BRD2, BRD3 and BRD4 in BMDM was sufficient to decrease the LPS-induced expression of interleukin (IL)6, IL1β, TNFα and monocyte chemoattractant protein-1.10 11 The expression of these genes was also significantly decreased in LPS-stimulated BMDM derived from brd2 lo mice, a genetically modified mouse model with a hypomorph phenotype and a 50% reduction of BRD2 expression in all tested tissues.11
Chromatin immunoprecipitations (ChIP) of TNFα and IL6 promoter regions in BMDM identified first gene-specific and distinct mechanisms behind BRD2 and BRD4 functions. Whereas BRD2 and BRD4 bound to the IL6 promoter, the TNFα promoter was bound only by BRD2 but not by BRD4. Since silencing of BRD4 was sufficient to decrease the TNFα secretion in LPS-stimulated BMDM, a chromatin-independent effect of BRD4 was suggested by the authors.11 Some of the anti-inflammatory effects of BET inhibitors might also be mediated by a direct effect of BET inhibitors on the activation of NF-κB pathways by decreasing the TNFα-induced phosphorylation of NF-κB inhibitor α (IκBα).12 66 However, these effects were not consistently observed and might be cell-type dependent or related to the treatment protocol and inhibitor used.10 15 60
BET protein inhibition in synovial fibroblasts
Besides the anti-inflammatory role of BET inhibitors in macrophages, I-BET151 and JQ1 were also sufficient to decrease the TNFα-induced, IL1β-induced, as well as the Toll-like receptor ligand-induced inflammatory response of SF and to decrease SF proliferation.15 60 61 The changes in gene expression of cytokines and chemokines translated into a reduced chemoattractance towards peripheral blood mononuclear cell.15 Silencing of BRD2 or BRD4 was equally efficient in downregulating the TNFα-induced expression of matrix metalloproteinases (MMP) and cytokines in SF.60 In addition, BRD4 was shown to control migration and invasion of SF.67 Similar to macrophages, only few target genes of individual BET proteins are known in SF. Recently, a study by Nagpal et al provided new insights into the mechanisms behind the activity of JQ1 in SF. JQ1 was shown to reduce the occupancy of both BRD2 and BRD4 in approximately 2000 regions and RNA Pol II in approximately 600 regions genome-wide in IL1β-treated SF. The authors showed that JQ1 reduced the BRD2/BRD4 occupancy in the IL6 and IL8 super enhancer regions in SF,61 consistent with a general function of BET proteins on super enhancers in different cell types.50 51 68 69
Besides the regulatory function of BET proteins in SF under inflammatory conditions, BET reading regulated the expression of HOX genes in a joint-specific manner under non-inflammatory conditions. I-BET151 decreased the basal expression of genes encoded in the 5′ HOXA cluster, but not of genes encoded in the 5′ HOXC cluster, which distinguish SF derived from upper versus lower extremity joints.70 This might be relevant to arthritis, since the expression of specific HOX genes widely overlaps with the occurrence of certain forms of arthritis in specific joints.71 As an exception among BRD proteins, BRD4 was shown to stick to the chromatin during mitosis in murine P19 embryonal carcinoma cells.38 This feature makes BET proteins to the key candidates that maintain the transcriptional memory in SF and other cells.
BET protein inhibition in endothelial cells
Endothelial cell activation and dysfunction are hallmarks of systemic autoimmune diseases such as rheumatoid arthritis, systemic lupus erythematosus (SLE) and systemic sclerosis (SSc).72–74 JQ1 treatment or silencing of BRD2 or BRD4 in human umbilical vein endothelial cells (HUVEC) suppressed the vascular endothelial growth factor (VEGF)-induced tube formation, vascular hyperpermeability and migration. The effects were accompanied with a reduced VEGF-induced phosphorylation of VEGF receptor 2 (VEGFR2), p21-activated kinase 1 and endothelial nitric oxide synthase, factors which are important for angiogenesis and permeability.75 In addition, BET BRD inhibition was shown to efficiently block the endothelial activation under inflammatory conditions. JQ1 or silencing of BRD2 and BRD4 in HUVEC cells suppressed the TNFα-induced or LPS-induced expression of adhesion molecules, translating into a reduced adhesion of leukocytes to activated HUVEC. Pretreatment of HUVEC with JQ1 suppressed the TNFα-induced phosphorylation and degradation of IκBα and reduced the nuclear levels of p65, indicating that the observed effects were at least partially due to a reduced activation of the NF-κB pathway.16 In line with this data, JQ1 decreased the p65 recruitment to IL6 and IL8 promoters in serum-stimulated human pulmonary microvascular endothelial cells (HPMEC), reducing the expression and secretion of IL6 and IL8. Furthermore, JQ1 reduced HPMEC proliferation, with increased expression of cyclin-dependent kinase (CDK) inhibitors CDKN1A and CDKN2D and decreased expression of CDK2, CDK4 and CDK6.76
BET inhibitors in mouse models of arthritis
As a proof of concept for the use of BRD inhibitors in RMD in vivo, in 2013, Mele et al showed that intraperitoneal injections of JQ1, over a period of 2 weeks, protected mice from collagen-induced arthritis (CIA), evident by reduced arthritis scores and histological assessment.34 These findings were later confirmed by two other groups.60 67 Recently, the use of a BDR2 specific and orally active BET inhibitor, RVX-297, was shown to be efficient in decreasing the arthritis scores in CIA mice and rats, as well as in mice with collagen antibody-induced arthritis.37 The beneficial effects of BET inhibitors in arthritis models in vivo were mainly attributed to the suppressive effects on Th17 cell differentiation and the reduced expression of Th17-associated cytokines, including IL17, IL21 and granulocyte macrophage-colony stimulating factor.34 37 The aberrant generation and activation of Th17 cells, a subset of T helper cells, is one of the key mechanisms leading to autoimmune conditions.53 JQ1 or a combined silencing of BRD2 and BRD4 also interfered with the differentiation of human Th17 cells and suppressed the expression of steroid receptor-type nuclear receptors (ROR)A and RORC, encoding the transcription factors RORα and RORγt that drive Th17 differentiation, as well as IL17A and IL21.34 These findings were later confirmed by differentiating Th17 from CD4+ T cells derived from healthy controls, patients with ankylosing spondylitis (AS) and psoriatic arthritis in the presence and absence of JQ1. In the same study, CBP30, a selective CBP/p300 inhibitor was described as a suppressant of human Th17 responses. In comparison to JQ1, CBP30 affected the expression of fewer genes in CD4+ T cells, but similarly downregulated the expression of RORC, IL17A and IL21.52 JQ1 was also shown to interfere with the RORC/IL17 pathway in imiquimod (IMQ)-induced skin inflammation, a mouse model for psoriasis-like skin disease. Topical application of JQ1 before and after IMQ-induced skin inflammation exhibited protective effects as indicated by reduced ear thickness, ear myeloperoxidase activity and decreased skin expression of RORC and IL17A.77
Mele et al showed that JQ1 had no impact on the differentiation of T cell subsets other than Th17 from human naive T cells, such as Th1, Th2 or regulatory T cells.34 In contrast, the administration of I-BET762, in early phases of T cell differentiation from naive murine T cells had long-lasting effects on the proinflammatory function of murine Th1 cells accompanied by an increase of anti-inflammatory genes such as IL10. Furthermore, I-BET762 did not affect the expression of RORC in this study, but was sufficient to reduce the expression of IL17A.33 In a doxicyclin-inducible mouse model for sustained and reversible silencing of BRD4 in vivo, the absence of BRD4 interfered with a normal haematopoiesis, leading to a depletion of CD4+ and CD8+ single-positive T cell subsets, as well as a significant reduction in Lineage−Sca1+cKit+ haematopoietic stem cells in reconstituted bone marrow.78 Differences in the results of the differentiation of different T cell subsets on BET inhibition might be due to different experimental setups or different inhibitors and inhibitor concentrations used. Bolden et al, for example, showed that the levels of BRD4 silencing (moderate vs strong) were essential in obtaining phenotypic changes in the intestine of mice.78
BET inhibitors in mouse models of SLE
Oral administration of JQ1 decreased the glomerular damage, nephritis, delayed the onset and decreased the severity of proteinuria in MRL-lpr mice, a murine model for SLE. The effects were accompanied by decreased serum concentrations of B-cell activating factor, IL1β, IL6, IL17 and IFNγ and augmented IL10. In addition, JQ1 treatment decreased serum concentrations of anti-dsDNA antibodies, as well as immune complex deposition in kidneys,36 a hallmark of human SLE. This is in line with the finding that BRD4 interacts with octamer-binding transcription factor 2 (Oct2), which is crucial for B-cell specific gene expression. JQ1 reduced the binding of Oct2 to the immunoglobulin kappa locus (IGK) promoter, leading to a BET inhibitor-mediated decrease in immunoglobulin production in B cells.59 In contrast to the study by Wei et al,36 the expression of LPS-induced IL10 was decreased in regulatory B cells (Breg) by JQ1 treatment.79 This might represent a detrimental effect of JQ1, since Breg cells are critically involved in suppression of inflammation and known to inhibit inflammatory immune diseases including CIA.80
BET proteins in bone homeostasis and cartilage metabolism
BET proteins have been suggested to play a role in bone homeostasis, which is regulated by bone-resorbing osteoclasts and bone-synthesising osteoblasts.81 I-BET151 and JQ1 suppressed the human and mouse nuclear factor κB ligand (RANKL)-induced osteoclastogenesis in vitro and in vivo.12 55 Therapeutic dosing of I-BET151 was sufficient to increase bone mass in postovariectomy osteoporosis. In K/BxN serum-induced arthritis, a mouse model resembling the inflammatory effector phase of arthritis, I-BET151 suppressed bone resorption, possibly by targeting osteoclast differentiation along with suppressing the ongoing inflammation. I-BET151 was shown to target nuclear factor of activated T cells C1, the master regulator of osteoclastogenesis by suppressing MYC.55 In the same study, the authors showed that I-BET151 could inhibit the differentiation of osteoblasts in vitro by using fivefold to 10-fold higher concentrations than those required for the suppression of osteoclastogenesis.55 In another study, I-BET151 inhibited the expression of MMP3, MMP9, RANKL and osteoprotegerin in human MG63 osteoblast cells treated with serum derived from patients with AS. The expression of these genes was also suppressed in HLA-B27/β2m transgenic AS Lewis rats, a rodent model for AS, on treatment with I-BET151.82 Unfortunately, no other outcomes of I-BET151 were studied in this animal model. Both, JQ1 and I-BET151 were shown to exhibit protective effects on cartilage, by downregulating the expression of major factors involved in extracellular matrix degradation in human, murine and rat chondrocytes.62 63 On surgical destabilisation of the medial meniscus (DMM), postsurgical treatment with I-BET151 from day 2 on prevented articular cartilage damage, but only at earlier and not at later time points after treatment.62 Whether this timely limited effect of I-BET151 in the DMM model was due to a general suppression of inflammation or other mechanisms was not further studied.
BET proteins in fibrosis
Fibrosis of the skin and internal organs, as well as vasculopathy are hallmarks of SSc.83 Transforming growth factor β (TGFβ) and downstream mothers against decapentaplegic homolog (SMAD) signalling molecules are key factors mediating the disease.84 Inhibition of BET proteins was efficient to inhibit lung,22 85 liver,19 renal23 and heart fibrosis20 in vivo.
Treatment of human dermal fibroblasts (DF) with JQ1 decreased the expression of alpha smooth muscle cell actin (αSMA), fibronectin, collagen (COL) 1A1, nicotinamide adenine dinucleotide phosphate oxidase (NOX)421 and TGFβ-induced cytokine secretion.85 Silencing of BRD4 in DF reduced stress fibre formation and resembled the effects on gene expression that were observed by JQ1 treatment at least partially.21 Therefore, some other BET proteins might additionally be involved in the beneficial effects mediated by JQ1 in DF. In pancreatic stellate cells, which are key regulators of the collagen I production and fibrosis in pancreatic ductal adenocarcinoma, silencing of BRD4 suppressed the expression of COL1A1 and COL1A2, whereas silencing of BRD2 or BRD3 led to opposite effects in this cell type. However, JQ1 was still sufficient to suppress collagen production in vitro and in vivo.86
In lung fibroblasts, both JQ1 and I-BET or silencing of either BRD2 or BRD4, decreased the TGFβ-induced expression of αSMA.22 Administration of JQ1 with the chow to mice with bleomycin-induced fibrosis prevented lung fibrosis.22 85 The expression of BRD4 was shown to be elevated in tubular and interstitial cells in humans with kidney disease, as well as in kidneys of mice with renal fibrosis induced by unilateral ureteral obstruction (UUO). Treatment of renal interstitial fibroblasts with I-BET151 or silencing of BRD4 reduced the TGFβ-induced expression of αSMA, fibronectin and COL1. In line with this, I-BET151 alleviated renal fibrosis in the UUO model.23 BET protein inhibition with JQ1 was also sufficient to suppress innate inflammatory and profibrotic transcriptional networks in mice subjected to transverse aortic constriction and in mice with permanent surgical ligation of the proximal left anterior descending coronary artery, resembling severe heart failure from prolonged pressure overload and massive anterior myocardial infarction, respectively. Together these data suggest beneficial effects of BET protein inhibition in multiorgan fibrosis and BRD4 as the major target in this context.
BRPF proteins in RMD
Studies on other BRD proteins than BET proteins in RMD are very rare. Recently, two studies on the role of BRD and plant homeodomain finger-containing (BRPF) family proteins were published. The BRPF family consists of three members: BRPF1, BRPF2 (BRD1) and BRPF3. BRPF proteins comprise several functional domains in addition to a BRD, including two plant homeodomains connected by a zinc finger and a PWWP domain.6 Pharmacological inhibition of BRPF proteins using the three pan-BRD-inhibitors PFI-4, OF-1 and NI-57 strongly impaired RANKL-induced differentiation of murine and human primary monocytes into bone resorbing osteoclasts accompanied by a decreased expression of osteoclast markers.56 In synovial tissues, BRPF2 expression was verified in SF and macrophages. BRPF2 exerts cell-type and stimulus-specific effects in SF and monocyte-derived macrophages (MDM). Silencing of BRPF2 in SF decreased proliferation and led to rather proinflammatory and prodestructive effects. In contrast, silencing of BRPF2 in human MDM exhibited mild anti-inflammatory effects, indicated by a decrease of the LPS-induced expression of TNFα.87 This data underscore the need for careful testing of BRD protein function and their inhibition in more than one cell type.
TRIM proteins in RMD
Another interesting group of BRD proteins is a subgroup of tripartite motif (TRIM) family proteins. TRIM proteins are multidomain proteins and are implicated in innate signalling pathways.88 Among TRIM proteins, TRIM28, TRIM33, TRIM24 and TRIM66 contain a BRD and are known as positive and negative transcriptional regulators.6 So far, no specific inhibitors targeting these TRIM proteins are available but an inhibitor with a dual activity targeting the BRD of BRPF1B and TRIM24 has been described.89 Knockdown of TRIM28 in human macrophages potentiated the IFN-regulatory factor 5-mediated expression of TNFα.90 Another study showed that TRIM33 is overexpressed in muscle and skin tissues of patients with dermatomyositis and skin tissues from patients with other autoimmune diseases including psoriasis.91 Studies in knockout mice revealed that TRIM33 acts as a transcriptional repressor in macrophages in vitro and in vivo, leading to a high sensitivity to endotoxin challenge. At early stages of haematopoiesis, TRIM33 deficiency was associated with an impaired production of monocytes/macrophages. Deficiency of TRIM33 in mature myeloid cells increased the expression of a subset of known LPS-repressible genes, whereas the expression of only few LPS-inducible genes was decreased.92 In addition, studies in mice showed that TRIM33 and TRIM28 play a crucial role in the differentiation of Th17 cells. The knockout of TRIM33 or TRIM28 in CD4+ T cells reduced the expression of IL17 and IL17F but had, in contrast to the BET protein-regulated inhibition of Th17 differentiation, no effect on Th17-related transcription factors such as RORC.57 58 These data point to different mechanisms underlying the BET protein-mediated and TRIM-mediated Th17 differentiation. In the absence of TRIM33 or TRIM28, the IL17 locus was enriched for suppressive histone marks, or reduced in active epigenetic marks, respectively, whereas the IL10 locus was enriched for activating H3K4me3 in TRIM33 knockout cells, leading to decreased and increased expression of the respective genes.57 58 TRIM33 orchestrated its effects by forming a transcriptional complex with RORγt and Smad2.57 On the other hand, a STAT3-dependent recruitment of TRIM28 was required for the RORγt recruitment and function at the IL17-IL17F locus.58
Conclusion and outlook
The development of small molecule inhibitors targeting BET proteins has raised new possibilities in the future treatment of RMD. Their effects among different mouse models are encouraging and raise the perspective that BET inhibitors might be used in the future for the treatment of different RMD including their comorbidities, such as cardiovascular events or chronic obstructive pulmonary disease. Preclinical and basic research studies provide indication of potential complications of BET inhibitor therapies, such as sterility, retroviral activation and impact on the haematopoietic development. The latter concern was confirmed by results of first phase 1 clinical studies in patient cohorts with lymphoma or multiple myeloma and patients with acute leukaemia (ClinicalTrials.gov, number NCT01713582). These studies reported reversible haematological toxicities, in particular thrombocytopenia, anaemia and neutropenia as the main related adverse events that were frequent among all dose levels. Few non-haematological events were reported, including gastrointestinal events and fatigue with increasing frequency and severity at the highest doses.93 94 Given that common BET inhibitors might be considered as non-hormonal approach to male contraception and also target the testis-specific BRDT protein, a BET protein essential for spermatogenesis, one of the adverse effects of BET inhibitor treatment could be male sterility.95–97 In addition, BET inhibitors are capable of reactivating latent retroviral gene expression.98 99 Although the exact role of retroviruses in RMD is incompletely understood, there is consensus about their role as triggers of autoimmunity 100 101 . Therefore, the use of BET inhibitors in patients with RMD might bear an unpredictable risk.
Ten years after the first publication of BET inhibitors, there is still remarkably little known on individual functions of single BET proteins. The majority of research has focused on the use of pan-BET inhibitors in different cell types in vitro and in mouse models in vivo and only few studies do a more in depth analysis on the underlying mechanisms of BET protein inhibition. Some studies have identified small numbers of target genes of selected BET proteins, mainly of BRD4, but full transcriptome analysis of BET proteins in different cell types and conditions are missing. Recent studies focused on the oral administration of BET inhibitors36 37 85 or the use of BRD2-selective inhibitors such as RVX-297.37 The good druggability of BRD will likely lead to the continuing development of inhibitors that will further boost the research in this field.
Studies on BRD proteins outside of the BET protein family are only appearing in the last few years and are still rare. Inhibitors targeting BRPF family proteins have only recently been developed 56 102 103 and will likely boost the research on BRPF proteins also in the field of RMD. So far, no TRIM protein-specific inhibitors are available for research, likely due to their dual function as activators and repressors of transcription. However, the interference of TRIM proteins with the differentiation of Th17 cells suggests that also TRIM protein inhibition might be a successful treatment in autoimmune conditions.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
Footnotes
Contributors KK planned and wrote the manuscript.
Funding The author has not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent Not required.
Provenance and peer review Commissioned; externally peer reviewed.
Data sharing statement No additional data are available.