Article Text

Abstract
Objective To investigate the efficacy, safety and immunogenicity of PF-06438179/GP1111 (PF-SZ-IFX) compared with European reference infliximab (Remicade®; ref-IFX) in patients with moderate-to-severe, active rheumatoid arthritis after continued long-term use of PF-SZ-IFX, and in patients who were switched from ref-IFX to PF-SZ-IFX.
Methods REFLECTIONS B537-02 was a double-blind, active-controlled, multinational study in which patients (N=650) were initially randomised to PF-SZ-IFX or ref-IFX for 30 weeks (treatment period [TP] 1). During weeks 30–54 (TP2), the PF-SZ-IFX group (n=280) continued treatment with PF-SZ-IFX (PF-SZ-IFX/PF-SZ-IFX) and patients in the ref-IFX group (n=286) were rerandomised (1:1) to continue ref-IFX (ref-IFX/ref-IFX) (n=143) or switch to PF-SZ-IFX (ref-IFX/PF-SZ-IFX) (n=143) for a further 24 weeks. Efficacy, safety, immunogenicity and pharmacokinetics were evaluated.
Results During TP2, patients in all three treatment groups continued to maintain comparable treatment response. At week 54, the American College of Rheumatology (ACR20) response rates were 71.1% (PF-SZ-IFX/PF-SZ-IFX), 64.3% (ref-IFX/ref-IFX) and 70.6% (ref-IFX/PF-SZ-IFX). Observations for other endpoints, including ACR50/70, Disease Activity Score in 28 Joints Based on High-Sensitivity C Reactive Protein(DAS28-CRP) remission, and mean change in DAS28-CRP and Health Assessment Questionnaire-Disability Index, were also comparable. Treatment-emergent adverse events were reported in 36.8% (PF-SZ-IFX/PF-SZ-IFX), 33.6% (ref-IFX/ref-IFX) and 37.8% (ref-IFX/PF-SZ-IFX) of patients; there were no clinically meaningful differences in the safety profiles between groups. The percentage of patients who were antidrug antibody-positive was generally stable through the treatment period and comparable overall between the PF-SZ-IFX/PF-SZ-IFX (52.1%; neutralising: 80.8%), ref-IFX/ref-IFX (60.1%; neutralising: 84.9%) and ref-IFX/PF-SZ-IFX (58.0%; neutralising 78.3%) groups.
Conclusions The similar efficacy, safety and immunogenicity of PF-SZ-IFX compared with ref-IFX were maintained for up to 54 weeks and were not affected by blinded treatment switch from ref-IFX to PF-SZ-IFX at week 30.
Trial registration number NCT02222493.
- anti-tnf
- dmards (biologic)
- rheumatoid arthritis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Treatment period 1 of the REFLECTIONS B537-02 study confirmed that PF-06438179/GP1111 (PF-SZ-IFX) has similar efficacy, safety and immunogenicity compared with European reference infliximab (ref-IFX) in patients with moderate-to-severe, active rheumatoid arthritis.
What does this study add?
These results from treatment period 2 of the same study continue to demonstrate similar efficacy, safety and immunogenicity of PF-SZ-IFX compared with ref-IFX after long-term follow-up and treatment switch from ref-IFX.
How might this impact on clinical practice?
These results add to the totality of evidence for PF-SZ-IFX and continue to support the use of PF-SZ-IFX as an infliximab biosimilar.
Introduction
Infliximab, a chimeric IgG1 monoclonal antibody that inhibits tumour necrosis factor alpha (TNFα), is a medicine for the treatment of several immune-mediated inflammatory diseases, including rheumatoid arthritis (RA), Crohn’s disease and ulcerative colitis.1 2
PF-06438179/GP1111 (PF-SZ-IFX) is a biosimilar of infliximab that is approved in the European Union,3 Japan4 and the USA.5 PF-SZ-IFX was developed by Pfizer. In February 2016, Sandoz acquired the biosimilar development, commercialisation and manufacturing rights from Pfizer for the 28 countries in the European Union plus Norway, Iceland and Liechtenstein. Pfizer retains commercialisation and manufacturing rights for the remaining countries of the world.
Biosimilars are cost-effective versions of their reference medicine that have undergone regulatory review and received regulatory approval based on a totality of evidence showing no meaningful difference compared with the reference medicine; thus, biosimilars are normally able to be approved for use in the same indications.6–8 Switching patients from reference medicines to biosimilars can provide cost savings9 10 and similar efficacy11–13; consequently, biosimilars offer the potential to expand patient access to important, but expensive, biologic medicines.14 Despite the potential benefits, there are still challenges and barriers to the uptake of biosimilars, including a desire for more information about biosimilars (eg, long-term efficacy and safety data) and concerns about the potential for increased immunogenicity after switching from a reference medicine to a biosimilar, or with multiple switching.14–16
To date, the totality of evidence demonstrates that PF-SZ-IFX has an identical amino acid sequence and similar biological activity to reference infliximab (Remicade; ref-IFX) as demonstrated in preclinical studies17; a similar pharmacokinetic profile in healthy volunteers to ref-IFX; and comparable immunogenicity with ref-IFX.18 The REFLECTIONS B537-02 study was a confirmatory phase III, double-blind, randomised, active-controlled trial.19 In treatment period 1 (TP1; weeks 0–30) similar efficacy, safety and immunogenicity of PF-SZ-IFX were shown compared with ref-IFX sourced in the European Union in patients with moderate-to-severe, active RA and inadequate response to methotrexate.19
Here, we report long-term data (up to 54 weeks) on the efficacy, safety and immunogenicity of PF-SZ-IFX compared with ref-IFX, including data for patients after a single switch from ref-IFX to PF-SZ-IFX after 30 weeks, from TP2 (weeks 30–54) of the REFLECTIONS B537-02 study.
Methods
Study conduct
An independent data monitoring committee was responsible for monitoring safety and study conduct. All patients provided written informed consent before entering the study. Since all three treatment periods were part of the same study, no additional informed consent was required prior to rerandomisation in TP2.
Study design and treatment
REFLECTIONS B537-02 was a multinational, randomised, double-blind, parallel-group, 78-week study comparing the efficacy, safety and immunogenicity of PF-SZ-IFX with ref-IFX (online supplementary figure S1).19
Supplemental material
The REFLECTIONS B537-02 study was split into three distinct treatment periods; TP2 (weeks 30–54) is the focus of this manuscript. As described previously, during TP1 (weeks 0–30), patients were randomised (1:1) to receive a 3 mg/kg intravenous dose of PF-SZ-IFX or ref-IFX at weeks 0, 2 and 6, and then every 8 weeks to week 30. At week 30, patients entered TP2; those receiving PF-SZ-IFX continued to receive PF-SZ-IFX (PF-SZ-IFX/PF-SZ-IFX group) and those receiving ref-IFX were blindly rerandomised (1:1), without stratification, to remain on ref-IFX (ref-IFX/ref-IFX group) or switch to PF-SZ-IFX (ref-IFX/PF-SZ-IFX group) for the following 24 weeks. Subject unique identifiers assigned at the screening visit were associated with patients’ randomisation and rerandomisation schedules and treatment assignments, and were retained throughout the study.
Patients continued to receive a stable dose of methotrexate (10–25 mg/week) and folic/folinic acid throughout the study. The investigators were asked to reconsider treatment in patients who did not show improvement, and continuation of treatment was at the discretion of the treating physician. TP3 was an open-label extension that assessed the longer term efficacy and safety of PF-SZ-IFX from weeks 54 to 78.
Patients
Briefly, the key inclusion criteria were male and female patients aged ≥18 years with moderate-to-severe, active RA (≥6 tender and ≥6 swollen joints, high-sensitivity C reactive protein level ≥10 mg/L, and on a stable dose of methotrexate [10–25 mg/week for ≥4 weeks]); a diagnosis of RA based on the 2010 American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) classification criteria for RA for ≥4 months; and class I, II or III of the ACR 1991 Revised Criteria for Global Functional Status in RA. Patients were excluded if they had current or prior treatment with infliximab or lymphocyte-depleting therapies (eg, rituximab, alemtuzumab); however, they were allowed ≤2 doses of one non-depleting, non-infliximab biologic if discontinued ≥12 weeks or five half-lives (whichever was longer) prior to the first dose of study drug. Further details have been described previously.19
Endpoints
The primary endpoint was the percentage of patients achieving ≥20% improvement in ACR response (ACR20) from study baseline (week 0), evaluated at week 14 during TP1, as previously reported.19
Secondary efficacy endpoints included ≥20%/50%/70% improvement in ACR response (ACR20/50/70) from study baseline; change from study baseline in Disease Activity Score in 28 Joints Based on High-Sensitivity C Reactive Protein (DAS28-CRP); DAS28-CRP remission, defined as a DAS28-CRP score <2.6; EULAR response; ACR/EULAR remission; change in tender and swollen joint counts from study baseline; high-sensitivity CRP and its change from study baseline; and Health Assessment Questionnaire-Disability Index (HAQ-DI) and its change from study baseline. During TP2, secondary endpoints were assessed at weeks 38, 46 and 54.
Safety and tolerability were evaluated throughout the study based on the reporting of adverse events (AEs), treatment-emergent adverse events (TEAEs) and adverse events of special interest (AESIs).
Immunogenicity was evaluated based on the number and percentage of patients in TP2 who had ≥1 postdose sample that tested positive for antidrug antibodies (ADAs); neutralising antibodies (NAb) were analysed in ADA-positive samples only. The methods used for evaluating immunogenicity are as previously described by Cohen etal.19
Statistical analyses
The study was powered (≥85%) to demonstrate equivalence using a prespecified symmetric margin of ±13.5% with a two-sided 95% CI when assuming ACR20 response rates of 57.5% at week 14 (TP1) in both arms.
The secondary efficacy endpoints were summarised using descriptive statistics, based on the intent-to-treat population, defined as all patients who were randomised to study treatment in TP2.
Safety and immunogenicity endpoints were analysed descriptively for the safety population (all patients who received ≥1 dose of study drug in TP2).
No formal hypothesis testing was conducted for any secondary, safety or immunogenicity endpoints, or for any endpoints measured during TP2.
Results
Patients and treatment
At study entry, 650 patients were randomised (PF-SZ-IFX, n=324; ref-IFX, n=326), as previously reported.19
At week 30, 566 patients who completed TP1 entered TP2. Of these, 280 continued PF-SZ-IFX treatment, and 286 patients treated with ref-IFX in TP1 were rerandomised to continue ref-IFX (n=143) or to switch to PF-SZ-IFX (n=143). Overall, 506 patients (89.4%) completed TP2 with similar rates of completion across the three treatment groups.
No clinically meaningful differences in baseline demographics were observed between the three treatment groups in TP2, or between those who entered TP2 versus TP1 (data not shown). Disease characteristics at week 30, prior to the first infusion of study medication in TP2, were not notably different between the three treatment groups but did reflect improvements from baseline at week 0 of TP1 (table 1).
Disease characteristics at week 30* (TP2 ITT population)
Efficacy
The primary endpoint was met in TP1 and has been reported by Cohen etal.19
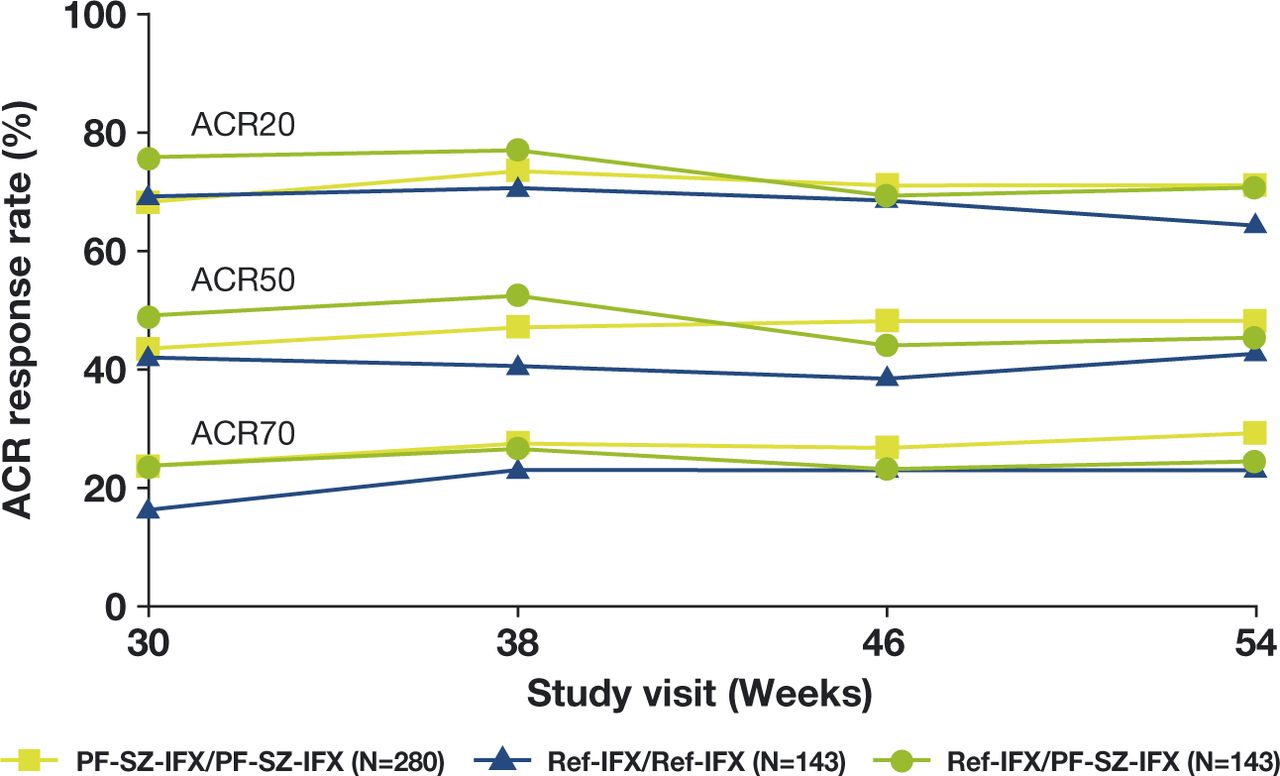
At week 30, prior to the first infusion of study medication in TP2, ACR20 response rates in the PF-SZ-IFX/PF-SZ-IFX, ref-IFX/ref-IFX and ref-IFX/PF-SZ-IFX groups, respectively, were 68.2%, 69.2% and 75.5%; ACR50 response rates were 43.6%, 42.0% and 49.0%, respectively; and ACR70 response rates were 23.6%, 16.1% and 23.8%, respectively. ACR20/50/70 response rates remained comparable between the three treatment groups at all study visits during TP2 (figure 1). At week 54, ACR20 response rates in the PF-SZ-IFX/PF-SZ-IFX, ref-IFX/ref-IFX and ref-IFX/PF-SZ-IFX groups were 71.1%, 64.3% and 70.6%, respectively; ACR50 response rates were 48.2%, 42.7% and 45.5%, respectively; and ACR70 response rates were 29.3%, 23.1% and 24.5%, respectively.

ACR20/50/70 responses during TP2 (TP2 ITT population). ACR20/50/70, ≥20%/50%/70% improvement in ACR response; ITT, intent-to-treat; PF-SZ-IFX, PF-06438179/GP1111; ref-IFX, European reference infliximab; TP2, treatment period 2.
At week 30 and prior to the first infusion of study medication in TP2, the mean DAS28-CRP value was 3.8 in all groups, reflecting mean changes from study baseline (week 0) of –2.2, –2.1 and –2.3 in the PF-SZ-IFX/PF-SZ-IFX, ref-IFX/ref-IFX and ref-IFX/PF-SZ-IFX groups, respectively. Throughout TP2, the mean change in DAS28-CRP from baseline remained comparable between groups at all TP2 visits (figure 2). At week 54, the mean DAS28-CRP values were 3.4, 3.6 and 3.6 in the PF-SZ-IFX/PF-SZ-IFX, ref-IFX/ref-IFX and ref-IFX/PF-SZ-IFX groups, corresponding to changes from study baseline of −2.5, –2.3 and −2.4, respectively.

Mean change in DAS28-CRP scores during TP2 (TP2 ITT population). DAS28-CRP, Disease Activity Score in 28 Joints Based on High-Sensitivity C Reactive Protein; ITT, intent-to-treat; PF-SZ-IFX, PF-06438179/GP1111; ref-IFX, European reference infliximab; TP2, treatment period 2.
The mean changes in HAQ-DI from baseline were comparable between groups at all TP2 visits (online supplementary figure S2). At week 30, the mean changes from study baseline in HAQ-DI were –0.6, –0.6 and −0.7 in the PF-SZ-IFX/PF-SZ-IFX, ref-IFX/ref-IFX and ref-IFX/PF-SZ-IFX groups, respectively, and −0.7, –0.6 and −0.8, respectively, at week 54. The mean changes in HAQ-DI from week 30 to week 54 were –0.03 (PF-SZ-IFX/PF-SZ-IFX), 0.02 (ref-IFX/ref-IFX) and –0.04 (ref-IFX/PF-SZ-IFX).
Overall, EULAR response rates, DAS28-CRP remission, mean tender/swollen joint counts and mean change in high-sensitivity CRP from baseline were comparable across the three treatment groups at weeks 30 and 54 (online supplementary table S1).
Safety
The median duration of treatment across all three treatment groups was 46.1 weeks between the first infusion of TP1 and the last infusion of TP2. No patient required a dose reduction in any group during TP2.
Treatment-emergent adverse events
Overall, the incidence of TEAEs and treatment discontinuations due to TEAEs during TP2 was low and comparable across the treatment groups (table 2). The most frequently reported TEAEs in the PF-SZ-IFX/PF-SZ-IFX, ref-IFX/ref-IFX or ref-IFX/PF-SZ-IFX groups during TP2 were infusion-related reactions (IRRs; reported in 3.2%, 8.4% and 4.2% of patients, respectively), nasopharyngitis (3.2%, 3.5% and 1.4% of patients, respectively) and exacerbation of RA (1.8%, 2.8% and 2.1% of patients, respectively).
Overview of treatment-emergent and treatment-related AEs during TP2 (TP2 safety population)
Treatment-related AEs
Overall, the incidence of TEAEs reported as potentially related to study treatment, including those leading to treatment discontinuation, was low and comparable across the three groups during TP2 (table 2). The most frequently reported treatment-related AEs in the PF-SZ-IFX/PF-SZ-IFX, ref-IFX/ref-IFX or ref-IFX/PF-SZ-IFX groups during TP2 were IRRs (3.2%, 7.7% and 4.2% of patients, respectively), rash (1.1%, 0% and 2.1%), dyspnoea (0%, 2.1% and 0.7%), nausea (0.4%, 2.1% and 0%), erythema (0%, 2.1% and 0%) and flushing (0%, 0% and 2.1%).
Adverse events of special interest
There were no clinically meaningful differences in AESI between the PF-SZ-IFX/PF-SZ-IFX, ref-IFX/ref-IFX or ref-IFX/PF-SZ-IFX groups in TP2, such as IRRs (3.2%, 8.4% and 4.2%, respectively), hypersensitivity (7.1%, 9.1% and 7.0%), overall infections (16.4%, 14.7% and 13.3%) and overall malignancies (0.4%, 0.7% and 1.4%).
The most frequently reported IRR TEAEs in TP2 were hypotension (1.1%, 0.7% and 0%), nausea (0.4%, 2.1% and 0%), pruritus (0.4%, 1.4% and 0%) and vomiting (0%, 1.4% and 0.7%). Across all treatment arms, very few serious (0.4%), grade 3 (0.7%) or grade 4 (0.2%) IRR TEAEs, and no grade 5 IRR TEAEs, were reported.
The most frequently reported infection-related TEAEs in TP2 were nasopharyngitis (3.2%, 3.5% and 1.4%), upper respiratory tract infection (2.1%, 1.4% and 2.1%), bronchitis (1.1%, 2.1% and 1.4%) and urinary tract infection (1.1%, 1.4% and 2.1%). Across all treatment arms, 1.1% reported serious infectious TEAEs and 1.4% of patients reported grade 3 or higher infectious TEAEs.
AEs leading to treatment discontinuation
Fourteen (5.0%), 10 (7.0%) and 7 (4.9%) patients in the PF-SZ-IFX/PF-SZ-IFX, ref-IFX/ref-IFX and ref-IFX/PF-SZ-IFX groups, respectively, permanently discontinued treatment during TP2 due to AEs. Discontinuations due to IRRs were reported in 6 (2.1%), 2 (1.4%) and 2 (1.4%) patients, respectively. Skin and subcutaneous tissue disorders led to 4 (1.4%), 1 (0.7%) and 3 (2.1%) patients, respectively, discontinuing treatment. Infections and infestations led to discontinuation in 2 (0.7%), 4 (2.8%; 2 patients due to latent tuberculosis [TB]) and 2 (1.4%; both due to latent TB) patients, respectively. All patients who discontinued due to latent TB had tested negative for TB during screening and experienced grade 1, non-serious latent TB events. Two cases of latent TB in the PF-SZ-IFX/PF-SZ-IFX group led to treatment discontinuation but were excluded from TP2 analyses by the TEAE programming cut-off (see online supplementary materials).
Immunogenicity
The percentages of patients who were identified as ADA-positive during TP2, regardless of ADA status in TP1, were comparable between the PF-SZ-IFX/PF-SZ-IFX, ref-IFX/ref-IFX or ref-IFX/PF-SZ-IFX groups (52.1%, 60.1% and 58.0%, respectively). Of those patients with a positive ADA test during TP2, 80.8%, 84.9% and 78.3% treated with PF-SZ-IFX/PF-SZ-IFX, ref-IFX/ref-IFX and ref-IFX/PF-SZ-IFX, respectively, also tested positive for NAbs.
The number of patients with ADAs and NAbs was comparable and generally stable over time between all three treatment groups (figure 3). A total of 288 patients (50.9%) in the TP2 population initially developed ADAs during TP1, and nearly all (n=270; 93.8%) continued to test positive for ADAs during TP2.

{kind=link}
{kind=link}
{kind=link}
ADAs and NAbs from weeks 30* to 54 (TP2 safety population). *Week 30 values were obtained prior to the first infusion during TP2. ADA positive and negative test results were defined as ADA titre ≥1.30 and <1.30, respectively. NAb positive and negative results were defined as NAb titre ≥0.70 and <0.70, respectively. ADA, antidrug antibody; NAb, neutralising antidrug antibody; PF-SZ-IFX, PF-06438179/GP1111; ref-IFX, European reference infliximab; TP2, treatment period 2.
As expected, ADA-positive patients had lower mean serum PF-SZ-IFX or ref-IFX trough concentrations than ADA-negative patients, but within each ADA subgroup the mean concentrations were generally comparable across the three treatment groups during TP2 (online supplementary table S2).
The majority of patients who developed ADAs had no hypersensitivity events and did not experience IRRs during TP2.
Of the 288 patients who were identified as ADA-positive in TP1, 32 experienced hypersensitivity TEAEs in TP2 (11 [7.1%], 13 [14.8%] and 8 [9.0%] in the PF-SZ-IFX/PF-SZ-IFX, ref-IFX/ref-IFX and ref-IFX/PF-SZ-IFX groups, respectively), and 25 experienced IRRs in TP2 (9 [5.8%], 10 [11.4%] and 6 [6.7%] in the PF-SZ-IFX/PF-SZ-IFX, ref-IFX/ref-IFX and ref-IFX/PF-SZ-IFX groups, respectively).
Of the 45 patients who were ADA-negative in TP1, but tested ADA-positive in TP2, only one patient in the ref-IFX/ref-IFX group reported an IRR on or after the date of first testing ADA-positive and none had experienced IRRs before testing ADA-positive. Two patients reported hypersensitivity TEAEs; however, these events occurred before these patients tested positive for ADAs.
Discussion
In line with the findings from TP1 of the REFLECTIONS B537-02 study,19 the present data from TP2 add to the totality of evidence supporting biosimilarity of PF-SZ-IFX compared with ref-IFX, and continue to demonstrate comparability between PF-SZ-IFX and ref-IFX in patients with moderate-to-severe, active RA. These data provide long-term evidence from a randomised, double-blind, controlled trial that the clinical efficacy, safety and immunogenicity of PF-SZ-IFX are comparable with that of ref-IFX up to 54 weeks, and that these factors were not affected by blinded treatment switch from ref-IFX to PF-SZ-IFX at week 30. These data provide additional reassurance to physicians who may want to see additional data compared with that required by regulatory agencies, and for those who have concerns about the potential for increased immunogenicity after switching from a reference medicine to a biosimilar.14–16
The primary endpoint of the study, ACR20 response assessed during TP1 at week 14, was met by similar percentages of patients treated with PF-SZ-IFX (62.7%) and ref-IFX (64.1%) and maintained until the end of the treatment period at week 30. At week 30 and after rerandomisation into the TP2 treatment groups, the ACR20 response rate remained comparable among patients who entered TP2, and these treatment responses were maintained for the duration of TP2 in all three treatment groups. Regulatory authorities consider ACR20 to be a sensitive efficacy endpoint in RA,20 and these findings therefore confirm continued comparable efficacy of PF-SZ-IFX and ref-IFX up to 54 weeks, including in those patients who were switched in a blinded manner from ref-IFX to PF-SZ-IFX at week 30. This finding is supported by other important measures of response in RA, including ACR50/70, EULAR good response, DAS28 change from baseline, DAS28-CRP remission and ACR/EULAR remission, all of which were maintained between weeks 30 and 54 in all three treatment groups. Slightly lower numerical response rates for ACR20/50/70 were observed in the ref-IFX/ref-IFX group compared with the other treatment groups, but these differences were not considered to be of clinical relevance by the study investigators. Small differences between the treatment groups may be explained, at least in part, because patients were not restratified at the beginning of TP2; however, differences in demographic and disease characteristics at the beginning of TP2 were not clinically relevant. In addition, differences in DAS28-CRP and HAQ-DI at any visit during TP2 were all less than the minimal clinically important difference of 1.021 and 0.22,22 respectively. Overall, the findings from TP2 provide long-term clinical data that complement the existing data demonstrating similarity of PF-SZ-IFX to ref-IFX, including identical amino acid sequences, structural similarity, similar biological function, pharmacokinetic similarity and therapeutic equivalence of PF-SZ-IFX compared with ref-IFX in patients with moderate-to-severe, active RA. This is supported by the fact that PF-SZ-IFX has been approved as a biosimilar of infliximab by regulatory bodies in the European Union,3 Japan4 and the USA.5
One of the current concerns with biosimilars is the potential for increased immunogenicity when switching from a reference medicine to a biosimilar of that medicine.19 Development of ADAs, particularly NAbs, is known to affect the pharmacokinetics of infliximab1 and other TNFα inhibitors, and could potentially impact efficacy and safety of the medication.23 Although this study was not powered to assess the impact of switching from ref-IFX to PF-SZ-IFX, the data collected do not provide any indication that switching had any meaningful impact on immunogenicity, pharmacokinetics, efficacy or safety. The percentages of patients who were ADA-positive and NAb-positive at week 30 were similar across all three treatment groups and remained comparable at week 54. Additionally, predose trough serum drug concentrations were generally comparable between the treatment groups throughout TP2. There were no apparent reductions in any of the measures of response between weeks 30 and 54 in any of the treatment groups, including those who switched from ref-IFX to PF-SZ-IFX, and there were no trends suggesting any differences in the safety profile of PF-SZ-IFX after treatment switch from ref-IFX. In clinical practice, it has been observed that switching from a reference medicine to a biosimilar can lead to treatment discontinuation or a switch back to the reference medicine due to reported loss of efficacy.24 25 It is notable that in this double-blind study, no loss of efficacy was observed and treatment retention rates were similar in TP1 (87.2%; based on safety population) and TP2 (89.4%) and comparable across treatment groups in TP2. This finding suggests that reported loss of efficacy in clinical practice is likely contributable to the ‘nocebo effect’ phenomenon,26 27 which could potentially be addressed, at least in part, by providing education for patients before making a shared decision between the treating physician and the patient, to switch to a biosimilar.28–31
Multiple biosimilars of infliximab have been approved in the European Union,3 32–34 USA5 35 36 and other countries. The evidence available to date is aligned with the findings presented here for PF-SZ-IFX, and suggests that there are no concerns relating to the long-term use of infliximab biosimilars, including after switch from ref-IFX.11–13 37–40
Study limitations
No formal hypothesis testing was conducted for any secondary endpoints and therefore no inferential statistics were generated. Study results were interpreted based on descriptive statistics.
Conclusions
TP2 (weeks 30–54) of the REFLECTIONS B537-02 study demonstrated sustained treatment effect of PF-SZ-IFX comparable with that of ref-IFX, and continued to show the absence of clinically meaningful differences in efficacy, safety and immunogenicity between patients with moderate-to-severe, active RA remaining on PF-SZ-IFX or ref-IFX, or after a randomised, double-blind switch from ref-IFX to PF-SZ-IFX. These results add to the totality of evidence and further support the biosimilarity of PF-SZ-IFX compared with ref-IFX.
Acknowledgments
Professional medical writing and editorial assistance was provided by Laura Maguire, MChem of Spirit, and was funded by Sandoz and Pfizer.
References
Footnotes
Contributors RA, BB, TH, HK, SCR, VT and SC contributed to the acquisition and interpretation of data. MR made substantial contributions to study conception and design, study conduct, and analysis and interpretation of data. MZ and CC contributed to the analysis and interpretation of data. GB, SH and OvR made substantial contributions to interpretation of data. All authors were involved in drafting the manuscript and/or revising it critically for important intellectual content, and read and approved the final manuscript for submission.
Funding This study was sponsored by Sandoz and Pfizer.
Competing interests RA has received honoraria and research grants from Bristol-Myers Squibb, Celltrion, Chugai Pharmaceutical, Eli Lilly, Janssen, Novartis, Roche and Pfizer. BB has received consulting fees, speaking fees and honoraria from Pfizer and Sandoz. TH has declared no conflicts of interest. HK has received consulting fees, speaking fees and/or honoraria from Asahi Kasei Pharma, Bristol-Myers Squibb, Chugai Pharmaceutical, Eli Lilly Japan KK, Janssen Pharmaceutical KK, Mitsubishi Tanabe Pharma, Novartis Pharma KK and Pfizer Japan, and research grants from AbbVie GK, Asahi Kasei Pharma, Astellas Pharma, Chugai Pharmaceutical, Eisai, Mitsubishi Tanabe Pharma, Novartis Pharma KK, Sanofi Pharma and UCB Japan. SCR has received research grants and consulting fees, speaking fees and/or honoraria from Pfizer. VT has no conflicts of interest. GB was an employee of Hexal (a Sandoz company) at the time of writing this manuscript. OvR is an employee of Hexal (a Sandoz company). CC, SH, MR and MZ are employees of Pfizer. SC has received consulting fees from Amgen, Boehringer-Ingelheim, Coherus, Merck, Pfizer and Sandoz and has received research grants from Amgen, Boehringer-Ingelheim, Coherus, Merck and Pfizer.
Patient consent for publication Not required.
Ethics approval The REFLECTIONS B537-02 study was conducted in accordance with the ethical principles of the Declaration of Helsinki and in compliance with all International Conference on Harmonisation Good Clinical Practice guidelines, and was approved by the independent ethics committee or institutional review board of each study centre.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Pfizer’s policies on the provision of clinical trial data are set out on its website: http://www.pfizer.com/research/clinical_trials/trial_data_and_results. In addition to posting clinical trial results on the ClinicalTrials.gov registry, Pfizer will provide access to anonymised patient-level data in response to scientifically valid research protocols. Data from Pfizer-sponsored global interventional clinical studies are available from trials conducted for medicines, vaccines and medical devices for indications that have been approved in the USA and/or by the European Union and from trials conducted for medicines, vaccines and medical devices that have been terminated (ie, development for all indications has been discontinued). Data from these trials will be made available 24 months after study completion. Pfizer will make reasonable efforts to fulfil all data requests for legitimate research purposes, but there may be instances in which retrieval or delivery of data is not feasible (eg, if Pfizer does not have legal authority to provide the data, if costs of retrieval of older or pre-electronic data are prohibitive; see page 5 at the following link: https://www.pfizer.com/files/research/research_clinical_trials/A_Guide_to_Requesting_Pfizer_Patient-Level_Clinical_Trial_Data_2017.pdf). Further details can be found at http://www.pfizer.com/research/clinical_trials/trial_data_and_results/data_requests. Pfizer’s practices adhere to the principles for responsible data sharing laid out by the European Federation of Pharmaceutical Industries and Associations (EFPIA) and the Pharmaceutical Research and Manufacturers of America (PhRMA); see http://phrma.org/sites/default/files/pdf/PhRMAPrinciplesForResponsibleClinicalTrialDataSharing.pdf.