Article Text

Abstract
Objectives To evaluate radiographic progression of structural joint damage over 2 years in patients with rheumatoid arthritis from baricitinib clinical trials who were disease-modifying antirheumatic drug (DMARD)–naïve or had an inadequate response to conventional synthetic DMARDs (csDMARD-IR).
Methods Patients had completed one of three phase III studies and entered a long-term extension (LTE) study, continuing on the same baricitinib dose as at originating study completion. At 52 weeks, DMARD-naïve patients receiving methotrexate (MTX) or combination therapy (baricitinib 4 mg+MTX) were switched to baricitinib 4 mg monotherapy (±MTX per investigator opinion); MTX-IR patients receiving adalimumab were switched to baricitinib 4 mg on background MTX. At 24 weeks, csDMARD-IR patients receiving placebo were switched to baricitinib 4 mg on background csDMARD. Radiographs at baseline, year 1 and year 2 were scored using the van der Heijde modified Total Sharp Score. Linear extrapolation was used for missing data.
Results Of 2573 randomised patients, 2125 (82.6%) entered the LTE, of whom 1893 (89.1%) entered this analysis. At year 2, progression was significantly lower with initial baricitinib (monotherapy or combination therapy) versus initial MTX in DMARD-naïve patients (proportion with non-progression defined by ≤smallest detectable change (SDC): 87.3% baricitinib 4 mg+MTX; 70.6% MTX; p≤ 0.001). In MTX-IR patients, progression with initial baricitinib was significantly lower than with initial placebo and similar to initial adalimumab (≤SDC: 82.7% baricitinib 4 mg; 83.5% adalimumab; 70.6% placebo; p≤0.001). In csDMARD-IR patients, significant benefit was seen with baricitinib 4 mg (≤SDC: 87.2% vs 73.2% placebo; p≤0.01).
Conclusions Treatment with once-daily baricitinib resulted in low rates of radiographic progression for up to 2 years.
- autoimmune diseases
- dmards (synthetic)
- rheumatoid arthritis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Three phase III clinical studies (RA-BEGIN, RA-BEAM, RA-BUILD) reported significant reductions in radiographic progression in baricitinib treatment groups for up to 1 year of treatment, including in patients who were disease-modifying antirheumatic drug (DMARD)–naïve or had previously failed to adequately improve on conventional synthetic DMARDs (csDMARDs) alone.
What does this study add?
The current analysis extends previous findings, demonstrating that treatment with baricitinib 2 and 4 mg maintains the inhibition of structural joint damage for up to 2 years in patients who completed one of the three original studies and entered the long-term extension study, RA-BEYOND.
How might this impact on clinical practice?
These data show that baricitinib, as monotherapy or in combination with csDMARDs, provides sustained structural benefits for up to 2 years in a broad spectrum of patients with rheumatoid arthritis (including DMARD-naïve patients and patients with an inadequate response to methotrexate and other csDMARDs).
Both 2 and 4 mg doses of baricitinib provided reduction on structural damage progression, with the most robust evidence seen for the 4 mg dose.
Introduction
Rheumatoid arthritis (RA) is a chronic, systemic autoimmune disease. Persistent inflammation can lead to structural joint damage, cartilage loss and erosive bone damage, which are associated with disability and poor patient outcomes.1 2 Current recommendations advise early treatment with disease-modifying antirheumatic drugs (DMARDs) to prevent or delay structural progression and the resulting impairments to function.3–5
Baricitinib is an oral, reversible inhibitor of Janus kinase (JAK)1 and JAK2, which mediate signal transduction for a variety of cytokines involved in inflammatory conditions, including RA.6 7 Baricitinib is approved for the treatment of moderately to severely active RA in adults in over 55 countries, including European countries, Japan and the USA. Three phase III studies previously evaluated the efficacy of baricitinib in terms of radiographic progression in patients with active RA who were DMARD-naïve or had an inadequate response (IR) to methotrexate (MTX) or to ≥1 conventional synthetic (cs)DMARDs.8–10 At weeks 24 and 52, a significant reduction in radiographic progression was observed with the combination of baricitinib 4 mg plus MTX compared with MTX alone in DMARD-naïve patients9 and with both baricitinib 4 mg and adalimumab on background MTX compared with placebo on background MTX in MTX-IR patients.10 Both baricitinib 2 mg and 4 mg were associated with significant inhibition of structural progression compared with placebo on background csDMARDs at 24 weeks in csDMARD-IR patients8 and at 1 year for patients who entered into a long-term extension (LTE) study.11 As RA requires long-term treatment, the objective of the current analysis was to extend previous findings, assessing radiographic and other efficacy outcomes following up to 2 years of baricitinib treatment for patients completing the originating phase III studies.
Methods
Patients
This analysis included patients who completed RA-BEGIN (DMARD-naïve: NCT01711359), RA-BUILD (csDMARD-IR: NCT01721057) or RA-BEAM (MTX-IR: NCT01710358) and entered into the LTE study RA-BEYOND (NCT01885078). The eligibility criteria for the originating studies have been previously described.8–10 In brief, eligible patients were ≥18 years old with moderately to severely active RA as defined by the presence of ≥6 tender and ≥6 swollen joints (from a 68/66-joint count) and a high sensitivity C-reactive protein (hsCRP) level ≥2× upper limit of normal (ULN) (≥6 mg/L) for RA-BEAM or hsCRP level ≥1.2× ULN (≥3.6 mg/L) for RA-BEGIN and RA-BUILD. All patients were biologic DMARD-naïve; additionally, patients were csDMARD-naïve or had an insufficient response or were intolerant to MTX or ≥1 csDMARDs.
Patients were eligible for RA-BEYOND if they had completed one of the originating studies. Rescue in the originating study did not preclude entry to RA-BEYOND. Patients were not eligible for participation if they were hypersensitive to baricitinib or if they permanently discontinued baricitinib during the originating study. Patients were also excluded if they had developed any significant medical issues or uncontrolled laboratory abnormalities during the originating study that were, in the opinion of the investigator, an unacceptable risk to the patient.
Study design
RA-BEYOND is an ongoing (study duration up to 7 years) phase III multicentre LTE of the safety and efficacy of baricitinib in patients who had completed prior baricitinib phase II and phase III clinical trials. The designs of the originating studies have been previously published8–10 (see figure 1 and online supplementary methods). Patients receiving blinded baricitinib at the conclusion of an originating study remained on that dose (2 mg or 4 mg, once daily) in RA-BEYOND in a manner blinded to the patient and investigator. In addition, the design of RA-BEYOND included a randomised, blinded substudy on dose step-down from baricitinib 4 mg to 2 mg in patients who had achieved sustained disease control on the higher dose (see online supplementary methods for more detail).12
Supplemental material
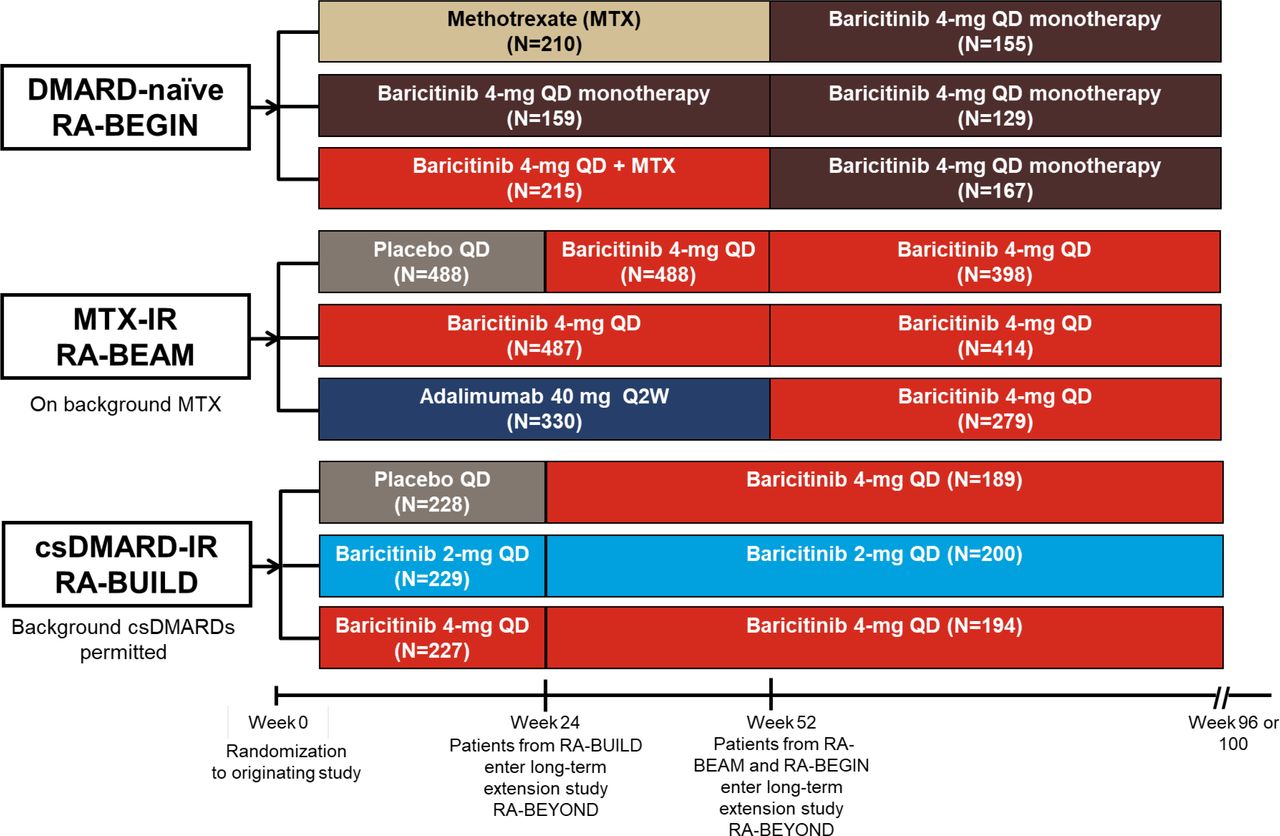
Design for the long-term extension study RA-BEYOND from randomisation in the originating studies. Data are up to 2 years on treatment, corresponding to 100 weeks in RA-BEGIN and RA-BEAM or 96 weeks in RA-BUILD due to the different duration and visit structures of the originating studies. Patients from RA-BEGIN switched to baricitinib 4 mg at entry to RA-BEYOND at week 52; during RA-BEYOND, MTX or other csDMARDs could be prescribed at any time point at the discretion of the investigators. Patients from RA-BEAM originally randomised to placebo were switched to baricitinib 4 mg at rescue or week 24; patients randomised to adalimumab were switched to baricitinib 4 mg at rescue or entry to RA-BEYOND at week 52. Patients in RA-BUILD initially randomised to placebo switched to baricitinib 4 mg at rescue or week 24 at entry to RA-BEYOND. Patients who remained on baricitinib 2 mg in RA-BUILD continued on 2 mg in RA-BEYOND; those rescued from baricitinib 2 mg to 4 mg in RA-BUILD received baricitinib 4 mg in RA-BEYOND. Background csDMARDs were permitted in RA-BUILD and RA-BEAM. csDMARD, conventional synthetic DMARD; DMARD, disease-modifying antirheumatic drug; IR, inadequate responder; MTX, methotrexate; N, number of patients randomised and treated; Q2W, once every 2 weeks; QD, once daily; RA, rheumatoid arthritis.
At 52 weeks in the DMARD-naïve study (at entry to the LTE), patients receiving MTX or baricitinib 4 mg plus MTX were switched to baricitinib 4 mg monotherapy but could be prescribed MTX or other csDMARDs at the discretion of the investigator at any time point during the LTE. Patients who initiated or increased csDMARDs above the doses used in the randomised, controlled phase of the study were captured through an Interactive Web Response System as rescued. At 52 weeks in the MTX-IR study (at entry to the LTE), patients receiving adalimumab on background MTX were switched to baricitinib 4 mg plus MTX. At 24 weeks in the csDMARD-IR study (at entry to the LTE), patients receiving placebo were switched to baricitinib 4 mg on background csDMARDs. Patients from this study who were rescued from baricitinib 2 mg to 4 mg received baricitinib 4 mg in the LTE; patients who received baricitinib 2 mg in the originating study without rescue continued on 2 mg in the LTE. In the LTE, csDMARDs could be initiated and dosages increased as rescue therapy for patients from the MTX-IR and csDMARD-IR studies who had a Clinical Disease Activity Index score >10 at or after 3 months following enrolment into the LTE; patients with normal renal function on baricitinib 2 mg could be rescued to 4 mg. Use of biologics was prohibited in the LTE.
Radiographic progression
Radiographic progression of structural joint damage was determined by the change from baseline in the van der Heijde modified Total Sharp Score (mTSS)13 and its subcomponents, the erosion score and joint space narrowing score. In addition, the proportion of patients showing no progression was determined based on change from baseline mTSS (ΔmTSS) of the originating study, using thresholds of 0, 0.5 or the smallest detectable change (SDC).14
To derive the mTSS, radiographs of the hands/wrists and feet were scored, quantifying the extent of bone erosion and joint space narrowing for 44 and 42 joints, respectively, with higher scores representing greater damage. Radiographs were scored by two central readers blinded to chronologic order, patient identity and treatment group. The mean score obtained between the two readers was used in the analysis. Baseline initial radiographs taken during the originating study served as the baseline radiographs for comparison throughout the LTE. Results comprise radiographs analysed in the same read campaign that included baseline, 1 year, and either 2-year or early termination time points. Radiographs were taken at study discontinuation if the most recent radiograph was more than 12 weeks earlier.
Clinical efficacy outcomes
The proportion of patients achieving specific thresholds in clinical efficacy measures was assessed at 12, 24 (DMARD-naïve, MTX-IR and csDMARD-IR studies) and 52 weeks (DMARD-naïve and MTX-IR studies) during the originating studies and every 12 weeks in the LTE up to 2 years. The outcomes examined included a Simplified Disease Activity Index (SDAI) score of ≤11 and a minimal clinically important difference (MCID) of at least 0.22 decrease from baseline on the Health Assessment Questionnaire–Disability Index (HAQ-DI).
Statistical analyses
Randomised patients who received at least one dose of the assigned study drug were included in the analyses. The analysis population for radiographic progression comprised patients with radiographs at baseline and at least one radiograph from the LTE after the 1 year radiograph. A mixed model for repeated measures (MMRM) was used to analyse continuous variables with treatment, visit and treatment-by-visit-interaction as fixed categorical effects and baseline and baseline-by-visit-interaction as fixed continuous effects. A logistic regression model with treatment included as a factor was used to analyse categorical variables (proportion of patients with no radiographic progression (ie, the proportion of patients meeting one of the thresholds set for ΔmTSS ≤0, ≤0.5 or ≤SDC), SDAI, HAQ-DI). The SDC was computed based on week 0 to year 1 and to year 2 changes in mTSS scores assigned by two blinded readers.14
Structural data observed after rescue or step-down were included in the analysis, and patients were analysed based on original randomisation. Linear extrapolation15–20 was used to impute missing scores at 2 years for patients who discontinued the study early and had radiographs available at 1 year and the early termination visit. The remaining missing scores at 2 years, for which linear extrapolation was not possible due to missing radiographs, were handled by the MMRM analysis (for continuous variables) or by exclusion of patients from the analysis (for the categorical variable of radiographic non-progression). The between-group comparisons were assessed with a significance level of 0.05 (two-sided). For SDAI and HAQ-DI data, non-responder imputation was applied after discontinuation of study or study drug. The data cut-off date was September 1, 2016.
Results
Patient disposition
Patient disposition is shown in figure 2 according to the treatment groups in the originating studies. Overall, 2573 patients were randomised, and the majority (N=2125; 82.6%) entered the LTE. Of these, the requisite radiographic assessments were available for 1893 patients (89.1%; RA-BEGIN (DMARD-naive): N=410, RA-BEAM (MTX-IR): N=1009, RA-BUILD (csDMARD-IR): N=474) for this analysis. The proportions of patients from RA-BEGIN or RA-BEAM in the analysis population of the LTE completing through 48 weeks (ie, 100 weeks, or 2 years, of treatment; figure 2A,B) and from RA-BUILD completing through 72 weeks (ie, 96 weeks, or 2 years, of treatment; figure 2C) were high (>94%) and similar across the original treatment groups in each study. Overall, 13 (3.2%), 37 (3.7%) and 13 (2.7%) patients from the DMARD-naïve, MTX-IR and csDMARD-IR studies, respectively, discontinued from the study during the analysis period in the LTE. In general, there were no major differences in discontinuations across the original treatment arms. The most common reason for discontinuation was an adverse event (DMARD-naive: n=8 of 13 discontinuations, 62%; MTX-IR: n=20 of 37 discontinuations, 54.1%; csDMARD-IR: n=6 of 13 discontinuations, 46.2%). For patients discontinuing early, linear extrapolation was applied (when possible) at year 2 for mTSS (0.0%–6.6% of patients per group; see online supplementary table 1).
Supplemental material

Patient disposition after 2 years of treatment. Summary of the analysis population in RA-BEYOND for patients originally completing (A) RA-BEGIN, (B) RA-BEAM or (C) RA-BUILD. Disposition is shown for up to 2 years on treatment, corresponding to a total of 100 weeks in RA-BEGIN and RA-BEAM (52 weeks in the originating study plus 48 weeks in RA-BEYOND) and 96 weeks in RA-BUILD (24 weeks in originating study plus 72 weeks in RA-BEYOND) due to the different durations and visit structures of the originating studies. Patients on placebo, MTX or adalimumab in the originating studies switched to baricitinib 4 mg at rescue or at 24 or 52 weeks, as detailed in the Study design section. The analysis populations in each group were the patients with radiographic assessments at baseline of the originating study and at least one radiograph from RA-BEYOND after the 1-year radiograph. LTE, long-term extension; MTX, methotrexate; QD, once daily; RA, rheumatoid arthritis.
The demographic characteristics of the 2-year analysis populations in the LTE were generally similar across the treatment groups at baseline of the originating studies: the majority of patients were white (>55%) and female (>70%), with a mean age range of 47.5 to 52.9 years (table 1). Clinical characteristics at baseline were similar across the treatment groups of the originating studies, but some characteristics differed across studies due to differences in the eligibility criteria of the originating studies (table 1).
Baseline characteristics of patients in the 2-year analysis populations in RA-BEYOND
Structural progression
Across the patient populations, treatment with baricitinib was associated with low progression of structural joint damage. For the DMARD-naïve patients originating in RA-BEGIN, those on initial baricitinib 4 mg plus MTX showed significantly smaller mean changes from baseline mTSS and erosion scores than patients on initial MTX at both years 1 and 2 (p≤0.001; figure 3A); those on initial baricitinib 4 mg monotherapy also had significantly fewer erosions than patients on initial MTX (p≤0.05). In agreement with these findings, a greater proportion of patients who had received initial baricitinib therapies in the DMARD-naïve study had no radiographic progression compared with patients who had received initial MTX monotherapy. These differences reached statistical significance at both years 1 and 2 for the initial baricitinib 4 mg plus MTX group when any of the thresholds were used (figure 4; see online supplementary table 2) and for the baricitinib 4 mg monotherapy group when either the ΔmTSS ≤0 or ΔmTSS ≤0.5 thresholds were used (see online supplementary table 2). For MTX-IR patients originating in RA-BEAM, those on initial baricitinib 4 mg had statistically significantly lower radiographic progression at years 1 and 2 than patients on initial placebo, and similar progression to patients on initial adalimumab (figure 3B). Both the baricitinib 4 mg and initial adalimumab groups also showed similar proportions of patients with mTSS nonprogression using any of the thresholds, which were statistically significantly higher than the proportion of patients showing no radiographic progression in the initial placebo group at both years 1 and 2 (figure 4; online supplementary table 2). For csDMARD-IR patients who had completed RA-BUILD, those on initial baricitinib 4 mg showed significantly smaller mean changes in mTSS and erosions (p≤0.05; figure 2C) and a greater proportion showed no radiographic progression (figure 4; online supplementary table 2) compared with patients on initial placebo at years 1 and 2 (p≤0.05; figure 2C). Progression with initial baricitinib 2 mg was numerically lower than with initial placebo (figure 3C); the proportion of patients showing no mTSS progression was also higher in the baricitinib 2 mg group compared with the placebo group at both 1 and 2 years, but these differences did not reach statistical significance.
Supplemental material

Inhibition of radiographic progression of structural joint damage at year 1 and year 2 by original randomisation. The LS mean change from baseline (±SEM) in structural joint damage evaluated using (A) mTSS, (B) ES and (C) JSN for patients in RA-BEYOND originally completing RA-BEGIN, RA-BEAM or RA-BUILD. Time points represent time from randomisation in the originating studies. Indicated treatment groups are according to randomisation in the originating study (‘initial’ denotes initial randomised treatment group). Patients on PBO, MTX or ADA in the originating studies switched to baricitinib 4 mg at rescue or at 24 weeks (PBO; single arrow) or 52 weeks (MTX and ADA; double arrow) as detailed in the Study design section. Tables represent the number of patients for whom data were available for each time point and study. Comparisons were analysed using a mixed model for repeated measures. Missing scores at 2 years were imputed using linear extrapolation based on data collected between 1 and 2 years. SE of the mean *p≤0.05, **p≤0.01, ***p≤0.001 baricitinib 4 mg vs PBO (RA-BEAM; RA-BUILD) or MTX (RA-BEGIN); +p≤0.05, ++p≤0.01, +++p≤0.001 ADA vs PBO (RA-BEAM); ‡p≤0.05 baricitinib 4 mg vs baricitinib 4 mg plus MTX (RA-BEGIN). ADA, adalimumab; Bari, baricitinib; ES, erosion score; JSN, joint space narrowing; LS, least squares; mTSS, modified Total Sharp Score; MTX, methotrexate; PBO, placebo; RA, rheumatoid arthritis.

Patient-level radiographic progression of structural joint damage at year 1 and year 2 by original randomisation. Panels show radiographic progression in structural joint damage evaluated using cumulative percentile change in the modified Total Sharp Score (mTSS) from baseline at year 1 and year 2 for patients in RA-BEYOND originally completing (A) RA-BEGIN, (B) RA-BEAM or (C) RA-BUILD. Each point represents an individual patient. Indicated treatment groups are according to randomisation in the originating study (‘initial’ denotes initial randomised treatment group). Inserted tables show n and percentage non-progression as defined by ≤SDC. Patients on placebo, MTX or adalimumab in the originating studies switched to baricitinib 4 mg at rescue or at 24 or 52 weeks, as detailed in the Study design section. **p≤0.01, ***p≤0.001 vs placebo (RA-BEAM; RA-BUILD) or MTX (RA-BEGIN). Δ, change from baseline; mTSS, modified Total Sharp Score; MTX, methotrexate; n, number of patients meeting threshold; N, number of patients with non-missing baseline and ≥1 non-missing post-baseline mTSS data; N-obs, number of patients included in analysis; RA, rheumatoid arthritis; SDC, smallest detectable change.
For all treatment groups, lower rates of change in mTSS were observed between years 1 and 2 (when all patients were on baricitinib therapies) than between baseline and year 1 (figure 3). Patients originally randomised to MTX or placebo exhibited greater increases in mTSS between years 1 and 2 (0.35±0.10 in RA-BEGIN; 0.56±0.09 in RA-BEAM; 0.33±0.09 in RA-BUILD) than those originally randomised to baricitinib treatment groups (0.21±0.01 and 0.24±0.09 for baricitinib 4 mg and baricitinib 4 mg plus MTX, respectively, in RA-BEGIN; 0.32±0.08 for baricitinib 4 mg in RA-BEAM; 0.28±0.09 and 0.24±0.09 for 2 and 4 mg, respectively, in RA-BUILD) (figure 3). Individual patient-level changes in radiographic progression from baseline were in accordance with these data at years 1 and 2 (figure 4).
Clinical efficacy
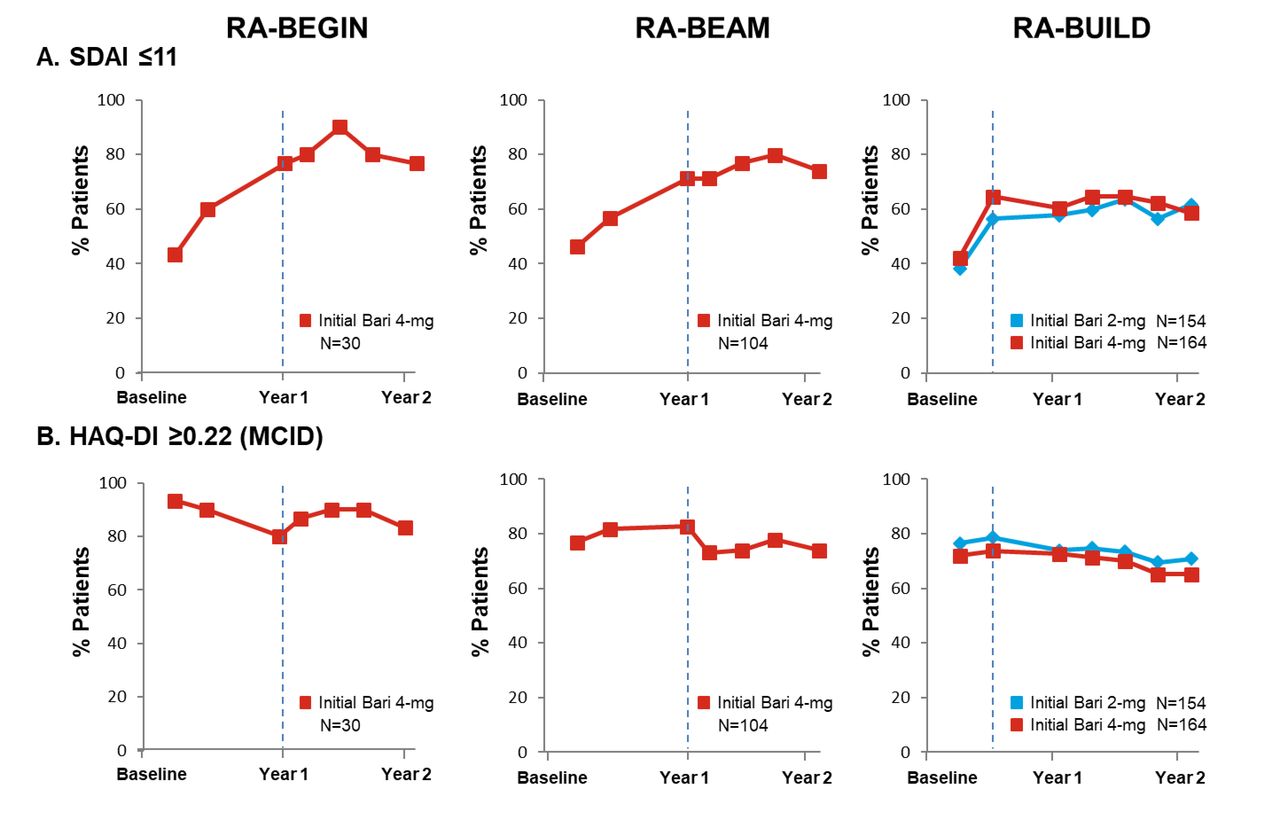
The majority (>60%) of patients across studies receiving baricitinib treatment achieved SDAI ≤11 at entry into the LTE study RA-BEYOND (figure 5A), which was sustained for up to 2 years of treatment. Similarly, the majority of patients on baricitinib 4 mg across the studies had functional benefit from treatment, with approximately 75% of patients on baricitinib who met or exceeded the HAQ-DI MCID (≥0.22) by week 12 in the originating studies (figure 5B). More than 60% of patients across studies met or exceeded the HAQ-DI MCID (≥0.22) at both years 1 and 2. The patients receiving the 2 mg dose through 2 years showed similar results to those receiving the 4 mg dose in terms of both SDAI ≤11 and HAQ-DI MCID (≥0.22) response.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proportion of patients (NRI) on baricitinib achieving SDAI ≤11 and meeting or exceeding HAQ-DI MCID (≥0.22) up to year 2 by original randomisation groups. Baseline in the originating study was used in the calculation of response. Time points represent time from randomisation in originating study, with year 1 and year 2 corresponding respectively to week 52 and week 100 for RA-BEGIN and RA-BEAM and to week 48 and week 96 for RA-BUILD due to the different durations and visit structures of the originating studies. Time points between year 1 and year 2 are in 12-week intervals. Dotted lines indicate entry into the long-term extension study. Analyses exclude patients who were rescued in the originating studies. NRI was applied without regard to rescue status in RA-BEYOND. Data after patients stepped down to baricitinib 2 mg were imputed using the response rate of the patients who continued with baricitinib 4 mg. ‘Initial’ denotes initial randomised treatment group. Bari, baricitinib; HAQ-DI, Health Assessment Questionnaire–Disability Index; MCID, minimally clinically important difference; N, number of patients not rescued in the originating study, who continued on the same dose in RA-BEYOND and entered RA-BEYOND 96 weeks before the cut-off date; NRI, non-responder imputation; SDAI, Simplified Disease Activity Index; RA, rheumatoid arthritis.
Discussion
The current analysis is a report on radiographic outcomes in the LTE study, RA-BEYOND, including patients from three studies with active RA who were either DMARD-naïve or had an inadequate response to MTX or ≥1 csDMARDs. The key finding is that baricitinib inhibited structural progression for up to 2 years of treatment, as assessed by total mTSS, erosions and joint space narrowing. Patients initially started on MTX or placebo exhibited significantly larger changes from baseline mTSS than those started on baricitinib 4 mg, either alone or in combination with MTX, with a higher proportion of patients on initial baricitinib therapies exhibiting non-progression at years 1 and 2. Further, patients on initial baricitinib therapies exhibited minimal changes in mTSS between years 1 and 2 compared with those on initial placebo or MTX, indicating that the inhibition of structural progression associated with initial baricitinib persisted during the second year of treatment. In MTX-IR patients completing RA-BEAM, radiographic outcomes at 1 year were similar between patients initially randomised to baricitinib 4 mg and adalimumab and remained similar at 2 years (ie, 1 year following the switch of initial adalimumab-treated patients to baricitinib 4 mg). These data are consistent with, and further extend, the original findings of the three studies, which reported significant reductions in radiographic progression in the baricitinib treatment groups at the end of the randomised study periods8–10 and at 1 year for the patients who had completed the csDMARD-IR study RA-BUILD and entered the LTE.11 Collectively, the data show that baricitinib, as a monotherapy or in combination with MTX/csDMARDs, provides sustained structural benefits in many patients with active RA, including those who have previously failed to adequately improve on MTX or other csDMARDs. Further, the data indicate that patients originally randomised to placebo or MTX and then switched to baricitinib 4 mg at 24 weeks continue to exhibit worse structural progression than patients originally randomised to baricitinib, even after 2 years of treatment, indicating that early intervention with baricitinib therapies is beneficial in the long term.
For the current analysis, radiographs at baseline and 1 year were reread during scoring of the year 2 radiographs. Consistent with previous studies which showed statistically significant structural benefit of baricitinib 2 mg and 4 mg at 24 and 52 weeks,8 11 the current study demonstrates significant benefit with initial baricitinib 4 mg in this population with up to 2 years of treatment. In addition, numerically greater reductions in the mean change from baseline mTSS and a higher proportion of patients showing no mTSS progression were observed in patients treated with baricitinib 2 mg compared with those on initial placebo who, at years 1 and 2, would have been treated with baricitinib 4 mg for 24 and 72 weeks, respectively. Collectively, the data indicate that both doses are effective at reducing structural progression with the best outcomes observed when treatment was started sooner and a more robust structural benefit achieved with baricitinib 4 mg.
In keeping with the radiographic progression findings, the majority of patients across the three originating studies receiving baricitinib therapies achieved SDAI ≤11 and HAQ-DI MCID by entry to the LTE, and this was maintained for up to 2 years on treatment. These data suggest that the sustained improvements in disease activity observed with long-term baricitinib treatment were associated with structural benefit and improved function. Safety was not examined in the current report as separate assessments of the safety of long-term baricitinib treatment have been conducted recently, which reported no significant new safety signals during the LTE up to 288 weeks.11 21 22 Notably, a recent comprehensive safety report has been published, which used the same data cut-off date as the current paper and large integrated data analysis sets from the baricitinib clinical programme.22 Across anaysis sets, infections were the most common adverse events reported in the baricitinib groups, but no notable differences were observed across treatment groups in the rates of serious adverse events including deaths, permanent discontinuations due to adverse events, malignancies, major adverse cardiovascular events and serious infections.22 Collectively, the data indicate that baricitinib treatment has a long-term clinical benefit with an acceptable safety profile.
There are some limitations to this analysis. Although this study included a large patient population, representing both DMARD-naïve and csDMARD-IR patients with active RA, this analysis included patients with varying treatment regimens and exposures to baricitinib due to differences in the designs of the three originating studies and to rescue and taper during the LTE. The study design did not permit comparison between active treatments and doses across studies. In addition, the LTE was open label, and patients were aware that they were receiving baricitinib. However, both patient and investigator were blinded to original randomised treatment assignments and the dose of baricitinib. Further, all radiographs were scored by two central readers who were blinded to all clinical information.
In conclusion, treatment with once-daily baricitinib was associated with low rates of radiographic progression for up to 2 years of treatment. Patients on initial baricitinib showed significantly less progression than those starting with placebo or MTX then switched to baricitinib, and comparable progression with those starting with adalimumab then switched to baricitinib. These differences in the level of progression were observed at both years 1 and 2, indicating a persistent structural benefit with long-term baricitinib treatment. For csDMARD-IR patients, treatment with baricitinib 2 mg and 4 mg was associated with reduced radiographic progression compared with initial placebo, with baricitinib 4 mg providing more robust evidence than the 2 mg dose.
Acknowledgments
We thank the patients, their families and the study personnel who participated in these clinical trials. These data were first presented at the Annual European Congress of Rheumatology 2017 (EULAR Abstract FRI0087; conference paper in Annals of the Rheumatic Diseases 76(Suppl 2):510.2–511). The studies were sponsored by Eli Lilly and Company (Indianapolis, Indiana, USA) and Incyte Corporation. Eli Lilly and Company sponsored the production of this manuscript. Syneos Health provided medical writing (Kaye L Stenvers) and editing (Antonia Baldo and Rod Everhart) support.
References
Footnotes
Contributors DvdH, PE and MS designed the experiments and analysed and interpreted the data; LX and RAO analysed and interpreted the data; GM, MC, TI and YT interpreted the data. All authors assisted in drafting and/or revising the manuscript, contributed to its intellectual content and approved the final version to be submitted. The authors take full responsibility for the manuscript.
Funding The studies were sponsored by Eli Lilly and Company (Indianapolis, Indiana, USA) and Incyte Corporation. Eli Lilly and Company sponsored the production of this manuscript.
Competing interests DvdH: received consulting fees from AbbVie, Amgen, Astellas, AstraZeneca, BMS, Boehringer Ingelheim, Celgene, Daiichi, Eli-Lilly, Galapagos, Gilead, Janssen, Merck, Novartis, Pfizer, Regeneron, Roche, Sanofi, UCB and is director of Imaging Rheumatology bv. MS: received consulting and/or speaking fees from AbbVie, Bristol-Myers Squibb, Eli Lilly and Company, Johnson & Johnson, UCB. YT: received speaking fees and/or honoraria from Daiichi-Sankyo, Astellas, Eli Lilly and Company, Chugai, Sanofi, Abbvie, Pfizer, YL Biologics, Bristol-Myers, Glaxo-SmithKline, UCB, Mitsubishi-Tanabe, Novartis, Eisai, Takeda, Janssen, Asahi-kasei and has received research grants from Mitsubishi-Tanabe, Bristol-Myers, Eisai, Chugai, Takeda, Abbvie, Astellas, Daiichi-Sankyo, Ono, MSD, Taisho-Toyama. LX, GM, TI, MC, RAO: employees and stock owners of Eli Lilly and Company. PE: received grant/research support or consulting support from Abbott, AbbVie, Bristol Myers Squibb, Eli Lilly and Company, Gilead, MSD, Novartis, Pfizer, Roche, Samsung, Takeda, UCB.
Patient consent for publication Obtained.
Ethics approval All studies were conducted with the approval of an institutional review board and in accordance with the ethical principles of the Declaration of Helsinki of 1975 (revised in 1983) and the Good Clinical Practice guidelines.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. Lilly provides access to the individual patient data from studies on approved medicines and indications as defined by the sponsor-specific information (available at https://clinicalstudydatarequest.com). This access is provided in a timely fashion after the primary publication is accepted. Researchers need to have an approved research proposal submitted online (https://clinicalstudydatarequest.com). Access to the data will be provided in a secure data sharing environment after signing a data sharing agreement.