Article Text

Abstract
Compared with the original approved biological drug on which it is based, a biosimilar has highly similar physicochemical characteristics and biological activity, as well as equivalent efficacy and no clinically meaningful differences in safety and immunogenicity. Before they are approved, biosimilars must undergo a rigorous development process using state-of-the-art technologies to establish biosimilarity to the reference biological product. After approval, biosimilars must comply with good pharmacological practices for biological drugs. Several biosimilar disease-modifying antirheumatic drugs (bsDMARDs) based on the tumour necrosis factor inhibitors adalimumab, etanercept and infliximab have been approved for use in patients with rheumatic diseases. Substantial cost savings can be made if biological-naive patients begin treatment with bsDMARDs, and patients receiving original biological DMARDs (bDMARDs) are switched to bsDMARDs. Despite the consistently similar efficacy, safety and immunogenicity of bsDMARDs relative to their respective original bDMARDs, switching from a reference bDMARD to a bsDMARD can result in nocebo responses, such as subjective increase of disease activity and pain-related adverse events. This may have a negative impact on adherence to bsDMARDs in clinical trials and clinical practice. To ensure optimal and rational integration of bsDMARDs into rheumatology practice and realise the full cost-saving efficacy of these drugs, rheumatologists must be aware that careful communication of the cost-saving efficacy and safety of bsDMARDs to their patients is the key to a successful long-term switch to bsDMARD therapy.
- anti-tnf
- autoimmune diseases
- dmards (biologic)
- rheumatoid arthritis
- arthritis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Several biosimilar DMARDs (bsDMARDs) based on adalimumab, etanercept, infliximab and rituximab have been approved for use in patients with rheumatic diseases, and many more bsDMARDsare in the pipeline.
The European League Against Rheumatism (EULAR) recommendations discuss bsDMARDs in the context of health-economic aspects, and express a preference for lower cost therapies when there is similar efficacy and safety but, as with the original biologic DMARDs (bDMARDs), recommendations do not distinguish between approved bsDMARDs.
Despite the consistently similar efficacy, safety and immunogenicity of bsDMARDs relative to their respective original bDMARDs, switching from a reference bDMARD to a bsDMARD can result in nocebo responses, such as subjective increase of disease activity and pain-related adverse events
What does this study add?
This article reviews the relevant considerations and success factors for ensuring appropriate, rational integration of bsDMARDs into rheumatology practice.
Experience from one UK NHS Trust shows that the integration of bsDMARDs requires all stakeholders (clinicians, pharmacists, patients, etc) to have confidence in using biosimilars.
To avoid contributing to the nocebo effect, it is very important that clinicians carefully consider how they communicate with their patients, and make an effort to frame communications in a positive context.
Key messages
How might this impact on clinical practice?
Healthcare systems can make substantial savings if patients receiving reference biologic products are switched to biosimilars, and if biologic-naive patients are started on biosimilars rather than reference products, as long as the costs differ.
Cost savings from the use of bsDMARDs can be diverted to other aspects of management for these patients, thereby potentially improving the overall provision of care.
For bsDMARDs to be widely integrated into clinical practice, and for maximal cost savings to be achievedwith these drugs, all prescribers and patients need to be aware of the consistent efficacy and safety of bsDMARDs in relation to reference bDMARDs, aswell as their substantial cost benefits.
Introduction
Biological disease-modifying antirheumatic drugs (bDMARDs), such as monoclonal antibodies and receptor Fc-fusion proteins targeting tumour necrosis factor (TNF), are an important component of treatment for patients with rheumatic diseases.1–4 These bDMARDs improve outcomes in several rheumatic diseases and have significant efficacy in patients who do not have an adequate response to conventional synthetic DMARD therapy alone.5–8 Despite the ability of bDMARDs to improve the lives of many patients with rheumatic diseases, the high cost of these drugs limits widespread use and contributes to inequalities of care.1 9 10 The accessibility of bDMARD therapy for patients who could benefit from such treatment but cannot access it because of cost is expected to improve as lower cost agents become available.9 11 12
A range of bDMARDs is available for use in patients with rheumatic diseases, including five TNF inhibitors: the receptor-Fc fusion protein etanercept, the chimeric monoclonal antibody infliximab, the human monoclonal antibodies adalimumab and golimumab, and the PEGylated humanised Fab’ monoclonal antibody fragment certolizumab pegol.13 In addition to TNF inhibitors, bDMARDs with other mechanisms of action include abatacept (a Fc fusion protein targeting T-cell co-stimulation), rituximab (a chimeric monoclonal antibody targeting CD20+ B cells) and tocilizumab and sarilumab (monoclonal antibodies, humanised and human, respectively, targeting the interleukin-6 receptor).1 13 The European League Against Rheumatism (EULAR) does not distinguish between approved bDMARDs with respect to their efficacy, stating in recommendations for the management of rheumatoid arthritis that they can all be used without hierarchical positioning, unless specific contraindications exist.1
A biosimilar is a biological agent that contains a similar version of the active substance of an already approved original biological agent (reference product), and is intended to be used in the same manner as the reference product.14 15 Several biosimilar DMARDs (bsDMARDs) based on adalimumab, etanercept, infliximab and rituximab have been approved for use in patients with rheumatic diseases, and many more bsDMARDs are in the pipeline.11 12 EULAR recommendations have also addressed health economic aspects and expressed a preference for lower cost therapies when there is similar efficacy and safety.1 This will improve rational medicine use, defined as use of the most appropriate medicine, dose and duration of treatment to meet individual patient needs at the lowest possible cost to patients and their communities,16 in rheumatology practice.
In this article, issues relevant to ensuring appropriate, rational integration of bsDMARDs into rheumatology practice are reviewed.
The economic impact of TNF inhibitor bsDMARDS
The high costs of TNF inhibitors have secured their position as some of the greatest revenue-producing drugs in the world. Adalimumab (Humira), which had no direct competition from bsDMARDs in 2017, was the world’s best-selling drug, with US$18 billion in 2017 global sales for immune-mediated inflammatory diseases, while etanercept (Enbrel) and infliximab (Remicade), both of which had competition from bsDMARDs, had sales of about US$8 billion each.17 18
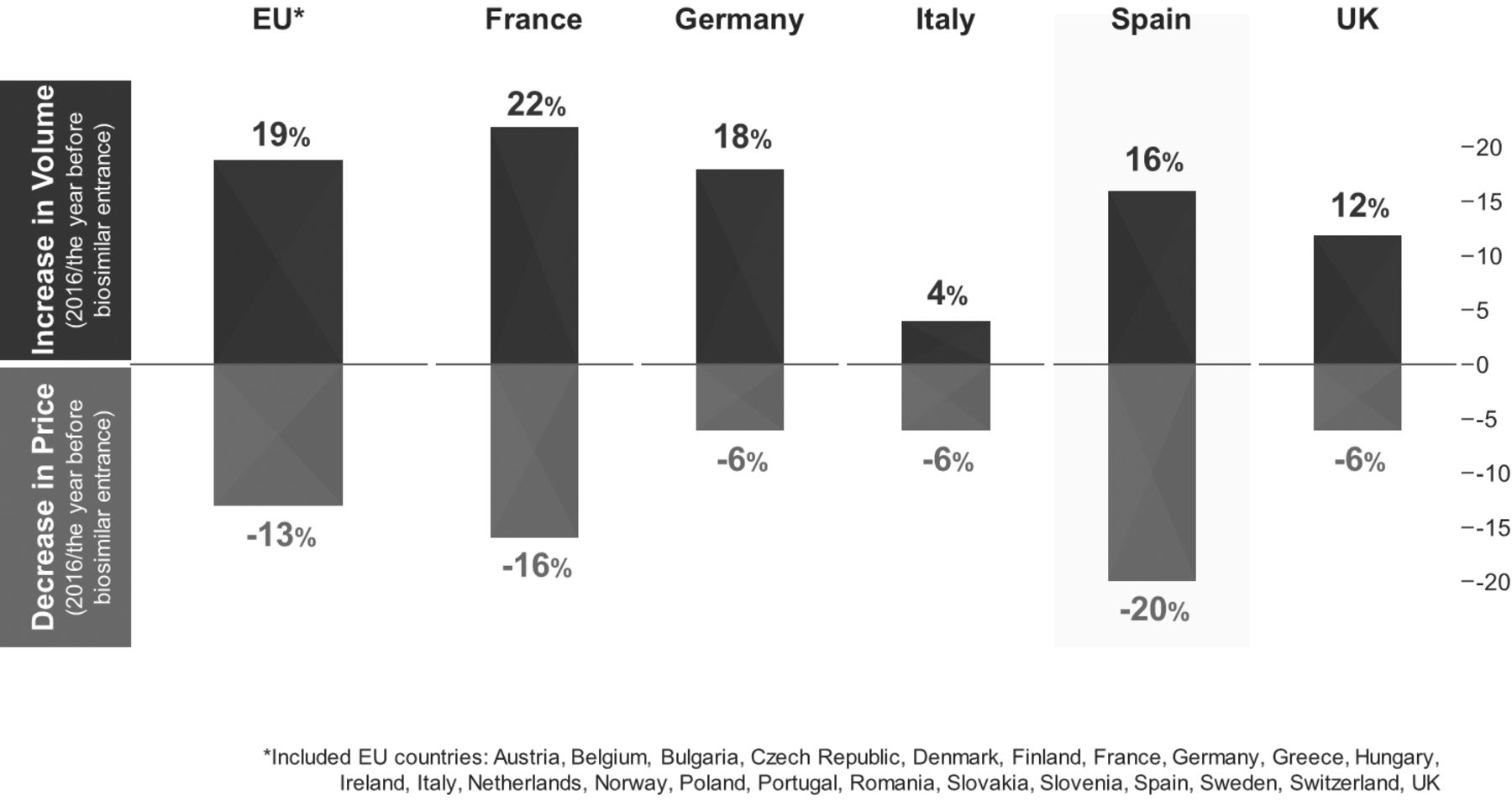
The infliximab biosimilars CT-P13 (Inflectra/Remsima)19 20 and SB2 (Flixabi),21 and the etanercept biosimilar SB4 (Benepali)22 were the first TNF inhibitor biosimilars to reach the European market (all before 2017). In 2016, the bsDMARD share-of-treatment days versus the reference product in the EU-5 ranged from 24.6% (France) to 64.1% (UK) for infliximab, and from 0.4% (Spain) to 31.6% (UK) for etanercept.23 Compared with 2016, the year before any etanercept or infliximab biosimilars was available on the European market, the price per treatment day across overall TNF inhibitor use decreased by 13% in the TNF inhibitor biosimilar accessible European Economic Area market, and volume per treatment day increased by 19% (figure 1).23 These changes indicate that etanercept and infliximab biosimilars are not only available at a lower cost per unit but are also facilitating access to these therapies for more patients. The launch of adalimumab biosimilars in the European Union (EU) in 2018 will further increase cost savings and the numbers of patients receiving effective bDMARD therapy.11 24
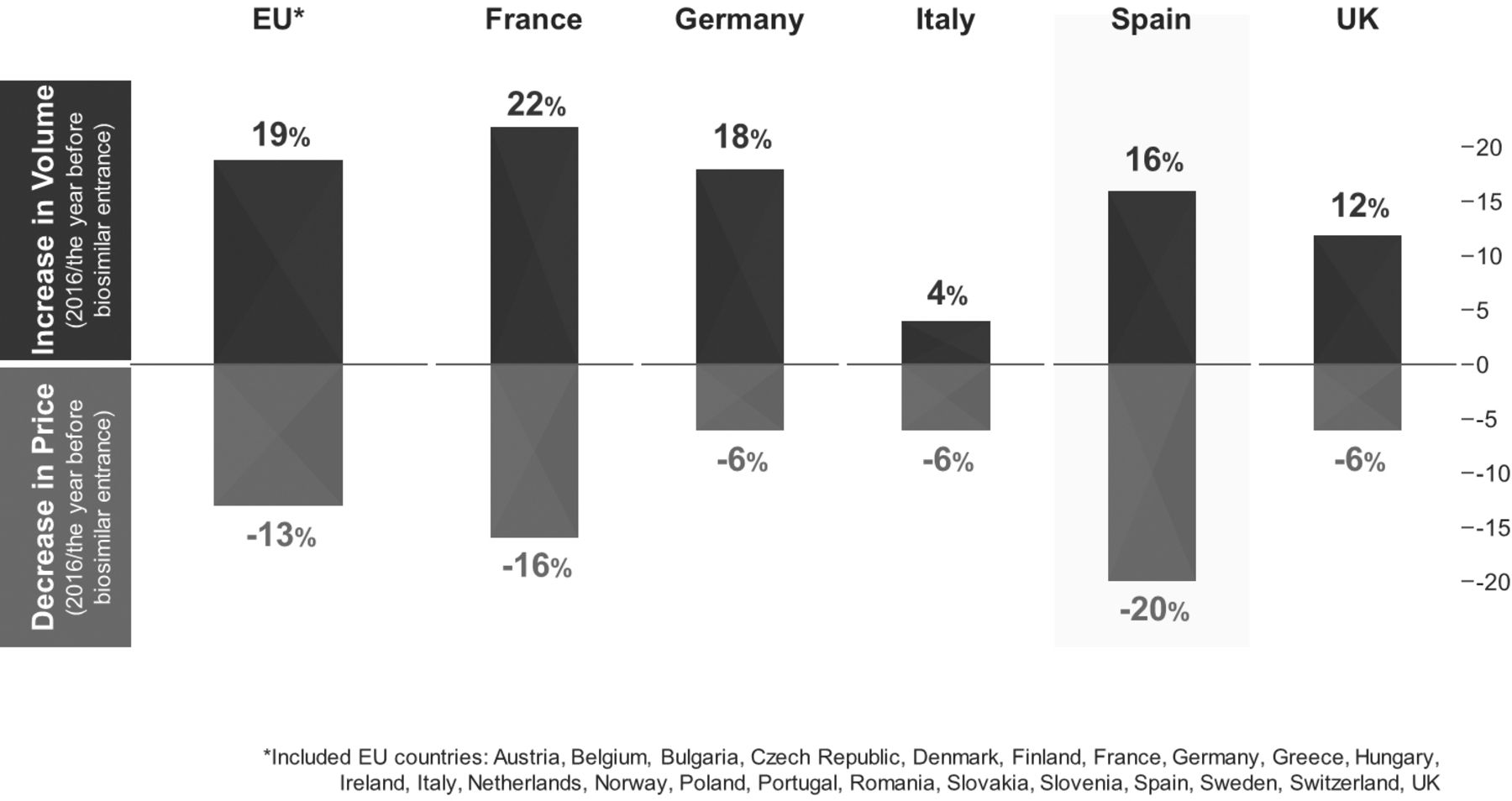
Health economic impact of anti-TNF biosimilars in the EU and EU-5 countries in 2016.23 EU, European Union; TNF, tumour necrosis factor.
The biosimilar development process
The European Medicines Agency (EMA) and the US Food and Drug Administration (FDA) require a biosimilar to have highly similar quality characteristics and biological activity to the reference product, with no clinically meaningful safety or efficacy differences.14 15 To get EMA or FDA approval, biosimilars must undergo a comprehensive development process that involves a series of comparability exercises to establish biosimilarity to the reference product, including at least one adequately powered randomised controlled trial (RCT) demonstrating equivalent efficacy of the biosimilar and reference product in an appropriate patient population.14 15 25 It is important to remember that these clinical equivalence studies are designed to optimise the chance of detecting a clinical difference between the biosimilar and the reference product in blinded homogeneous populations who have signed an informed consent to receive a biosimilar, and are therefore not reflective of biosimilar use in daily practice.26 27 As well as being assessed during the pivotal clinical efficacy study, clinical safety is initially assessed during clinical pharmacokinetic/pharmacodynamic studies.14 15
Compared with the development process for reference products, there is much more emphasis placed on physicochemical and functional characterisation of biosimilars than on clinical testing (figure 2).14 15 25 To ensure that there are no clinically meaningful differences between the biosimilar and the reference product, and thereby reduce the need for clinical testing, critical quality attributes (CQAs), which relate to physicochemical properties, and biological and immunological functions that could affect efficacy and safety, must be within prespecified quantitative target ranges that account for expected batch-to-batch variability and are therefore representative of the reference product.25 28 For example, the results of TNF neutralisation assays for anti-TNF biosimilars should be within the target range defined for the anti-TNF reference product.29 30 As shown in figure 3, this has been demonstrated during development of the etanercept biosimilar SB4 in relation to multiple batches of EU-sourced and US-sourced reference product, whereby the target range was set by a statistical analysis that generated a probability interval ensuring, within a specified confidence level, to contain a specified proportion of the population.29 For anti-TNF monoclonal antibody biosimilars, the results of assays reflecting TNF-binding, the corresponding neutralisation effect of that binding and effector functions associated with the Fc domain, such as C1q binding, complement-dependent cytotoxicity and antibody-dependent cell-mediated cytotoxicity, should also be within target ranges of the reference product.30 These examples are, however, only a subset of the comprehensive series of structural, physicochemical and biological analyses that are conducted to demonstrate similarity between anti-TNF biosimilars and reference products.29 30

The stepwise approach to the development of reference and biosimilar products.14 15 25

Comparison of TNF neutralisation activity of SB4 and etanercept reference product (40 lots of EU-sourced product and 40 lots of US-sourced product) measured using a TNF neutralisation assay that uses a luciferase reporter gene cell line containing an upstream NF-kB binding sequence. The dotted line indicatesthe similarity range (mean ± κSD) based on results of etanercept obtained from the EU.29 EU, European Union; TNF, tumour necrosis factor.
The aim of biosimilarity clinical trials is not to establish efficacy per se, which has already been established in clinical trials conducted with the reference product, but to demonstrate equivalent clinical performance of the biosimilar in relation to the reference product, with prespecified equivalence margins determined on the basis of data from previous RCTs with the reference product.14 When biosimilarity is demonstrated in one indication, this can be extrapolated to other approved indications of the reference product as long as the product’s mechanisms of action are consistent across the different indications.14 15 For example, several adalimumab, etanercept and infliximab biosimilars have been approved by the EMA for the full range of immune-mediated inflammatory indications of the reference products, with most clinical testing of these agents having been conducted in patients with rheumatoid arthritis.31 32
To test for potential changes in efficacy and safety after switching from a reference product to a biosimilar, some RCTs have incorporated a transition design, which is often an open-label extension component during which all patients receive the biosimilar.33 34 The adalimumab biosimilars SB5 and BI 695501 have recently been shown to have equivalent efficacy, and similar safety and immunogenicity to adalimumab in RCTs in patients with moderately to severely active rheumatoid arthritis despite treatment with methotrexate.26 33–35 These studies incorporated a double-blind transition design whereby, at week 24, patients who were initially randomised to receive reference adalimumab were rerandomised to continue with reference adalimumab or to switch to biosimilar adalimumab up to week 42 (BI 695501) or week 52 (SB5); while patients initially randomised to receive biosimilar adalimumab continued treatment.26 33 American College of Rheumatology response rates, which were equivalent between the SB5 and reference adalimumab treatment groups at week 24,35 were not affected by the switch from reference adalimumab to SB5 (figure 4).33 Efficacy was maintained and SB5 was well tolerated, with comparable safety to reference adalimumab, over the course of the study, and there was no increase in the incidence of antidrug antibodies after a switch from reference adalimumab to SB5.33 Similarly, the switch from reference adalimumab to BI 695501 did not have any negative effects on efficacy, safety or immunogenicity.26 Although the transition components of these studies were not designed for statistical comparisons of equivalence, they do provide valuable data on switching from a reference product to a biosimilar.26 33
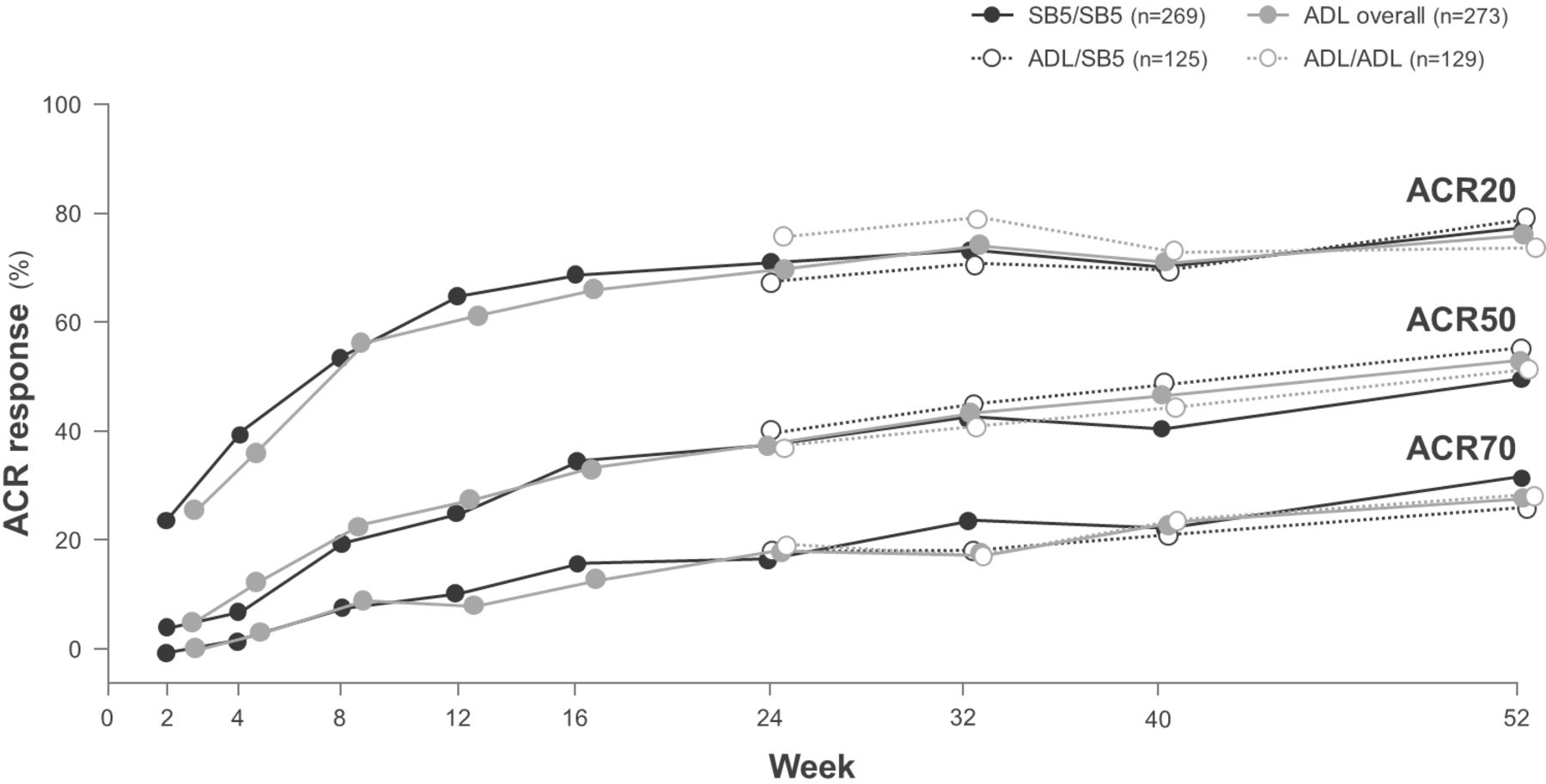
ACR20, ACR20 and ACR70 response rates (proportions of patients meeting ACR 20, 50% or 70% improvement criteria) in patients with rheumatoid arthritis randomised to receive double-blind SB5 (SB5 group) or reference adalimumab (ADL overall group) at week 0, and patients in the ADL group who were randomised at week 24 to continue double-blind ADL (ADL/ADL group) or switch to SB5 (ADL/SB5 group) up to week 52 in a phase III clinical trial.33 ACR, American College of Rheumatology; ADL, adalimumab.
To support a designation of interchangeability between a reference product and biosimilar, which includes switching (when a prescriber changes one drug for another) and substitution (when the change occurs at the pharmacy level without consultation with the prescriber), an RCT comparing patients who undergo multiple treatment switches with those continued on the same treatment should be conducted.12 36 To date, most RCTs have included only one treatment switch, but biosimilarity has also been demonstrated in clinical studies with multiswitch protocols.27 36 For example, in the EGALITY (study, patients with moderate-to-severe chronic plaque-type psoriasis were randomised to undergo a sequence of three treatment switches between GP2015 and reference etanercept. GP2015 demonstrated equivalent efficacy and comparable safety and immunogenicity to reference etanercept, and multiple switching between the two drugs did not impact safety or effectiveness.37 38 Similar findings were reported in another study of patients with moderate-to-severe plaque-type psoriasis who were randomised to multiple switches between GP2017 and reference adalimumab.39 As with the single-switch studies described above, the primary focus of these studies was to assess biosimilarity, and they were not powered to assess treatment switching.39 Thus far, no studies have been published that examine the effects of switching between different biosimilars of the same reference product, but such studies will become increasingly important to clinical practice as clinicians gain access to multiple biosimilars of the same reference medicine27 39
Reinvestigating biologics quality standards
After completion of the biosimilar development process, comparability testing that complies with good pharmacovigilance practices for biological drugs is performed during manufacturing process development and production scale-up, and after any changes to manufacturing process are made.40–43 Comparative prechange and postchange non-clinical and clinical data may be required.41 However, although biological products that undergo manufacturing changes are not necessarily identical to the originally approved drugs to the extent that they may in some cases be considered biosimilars of themselves,44 demonstration of pre and postmanufacturing change comparability of a biological product has historically required less sophisticated methods and less comprehensive characterisations than demonstration of biosimilarity.15 25 28 30 Ongoing comparability analyses using highly sensitive state-of-the-art technology reduce uncertainty about the efficacy and safety of biosimilars by demonstrating that CQAs are maintained within highly similar range of their reference products, and that any small changes in the biosimilar’s physicochemical profile that occur as a result of production process changes are not likely to adversely affect clinical safety or efficacy.41 43
In particular, when switching from a reference product to its biosimilar, there have been concerns about potential differences in immunogenicity resulting from structural differences between the two molecules.45 46 For example, aggregation of biological proteins increases the risk of immunogenicity.47 Any identified heterogeneities in the aggregation and other CQAs of a bsDMARD relative to its reference product must, therefore, be assessed for their potential impact on immunogenicity.25 As recently demonstrated in relation to epitope binding of CT-P13 and infliximab reference product, it is important to know that switching from a bDMARD to a bsDMARD is not likely to invoke any new immunogenic reactions.46
Using state-of-the-art technologies to monitor the physicochemical profile
The physicochemical profiles of approved biosimilars must be stable and consistently maintained. The availability of highly sensitive state-of-the-art analytical technologies allows recognition of potential effects on protein immunogenicity and small structural modifications that could affect the overall stability of the drug. These technologies are used in comparability exercises to detect differences in the CQAs of biosimilar and reference biological products, including primary and higher order protein structure, aggregation, glycosylation and charge heterogeneity.29 30 CQAs of the biosimilar are compared with multiple batches of reference product over an extended period of time since initial approval as possible, including pre and postmanufacturing change batches.28,28
For example, results of comprehensive comparability exercises using state-of-the-art analytical methods showed the overall physicochemical profile of the infliximab biosimilar SB2 to be highly similar to multiple batches of EU-sourced and US-sourced reference product, with no increased risk of immunogenicity.30 Neonatal Fc receptor binding activity, which can affect pharmacokinetics, was within the similarity range of the infliximab reference product for SB2.30 Clinical trial results in healthy volunteers confirmed SB2 had equivalent pharmacokinetics to infliximab reference product, as well as comparable safety and immunogenicity profiles.48 Results of comprehensive comparability exercises for SB4 and etanercept reference product showed the biological activity of SB4 was highly similar to the reference product in terms of TNF binding and neutralisation.29 CQAs that were out of range indicated that SB4 contained lower levels of undesirable impurities than the reference product.29 An RCT in patients with rheumatoid arthritis demonstrated equivalent efficacy for SB4 and etanercept reference product up to 52 weeks, but significantly fewer injection-site reactions and less immunogenicity was associated with SB4 than the reference product.49 50
Biosimilar adoption: improving rheumatology services for patients
Healthcare systems can make substantial savings if patients receiving reference biological products are switched to biosimilars, and if biological-naive patients are started on biosimilars rather than reference products, as long as the costs differ.9 In the UK, it has been estimated that as the biosimilar market develops over the next year, there is potential to realise savings of at least £200 million to £300 million per year by 2020/2021 through increased uptake of the best-value biological medicines.51 These savings can then be reinvested into other aspects of patient care and/or be used to increase patient access to biologics.9 11 51 52
Switching to a biosimilar: the National Health Service experience
In 2016, the York Trust Rheumatology Service instigated a switch process to manage the launch of the etanercept biosimilar SB4. The etanercept switch process planning phase coincided with positive position statements on biosimilars from the National Institute for Health and Care Excellence (NICE) and the National Rheumatoid Arthritis Society 53 54 and a growing body of data supporting switching to bsDMARDs.52 55
In their 2016 biosimilar statement, NICE recommended identifying clinical and pharmacy champions to take the lead in introducing biosimilars to clinical practice.53 It was also recommended that all stakeholders, including patients, should be consulted to ensure confidence in using biosimilars, extrapolation and equivalence of clinical data should be explained, and information about the EMA licensing process for biosimilars and the manufacturing process should be provided. Cost-saving and reinvestment opportunities should be identified, and gain-share agreements explored (arrangement between healthcare commissioners and providers to share the benefits associated with more efficient use of high-cost medicines that are directly commissioned by the National Health Service (NHS)).53 56 Once clinical consensus is established, approval for biosimilars should be sought from the local formulary committee. Data should be collected at baseline and after the introduction of biosimilars, and these data should be submitted to national audits and registries, such as the British Society of Rheumatology Biologics Register (http://www.bsrbr.org/) for pharmacovigilance purposes.53 All of these recommendations were considered in the etanercept switch planning process.
Based on annual predicted costs and a 50% gain-share agreement against the originator cost price, £1.64 million of extra income was forecast for the department over 2 years for the cohort switch (n=377) to the biosimilar. Notification letters and a ‘frequently asked questions’ leaflet were sent to the patients explaining regulatory authority approval of biosimilars, emphasising the efficacy and safety of biosimilars in patients with rheumatoid arthritis and the potential cost savings of switching, and offering an opportunity to receive further information if required (by telephone or face-to-face discussion). In surveys sent out after the switch, patients indicated that they particularly valued the written communication and the opportunity for face-to-face discussion.
After receiving information on the switch, 363 (96%) patients switched to biosimilar etanercept, and 14 patients (4%) declined switching. At 3 months, 97% of patients reported no change in disease activity, and 3% of patients had reverted to the reference product (lack of efficacy cited as the most common reason; two patients had skin rashes). At 18 months, 8% of patients had discontinued (6% due to lack of efficacy and 2% for adverse events).
As well as providing substantial cost savings to the NHS, the etanercept switch programme allowed the York Trust Rheumatology Service to fund additional nurse specialist time, two administration support roles and a biological pharmacist. The savings from the etanercept switch programme have facilitated better access to biologics for patients with non-funded indications, and for those who do not meet NICE eligibility criteria. As a result, more patients who may benefit from biological agents are now receiving them. In general, the etanercept switch programme improved the efficiency of the rheumatology service in terms of administrative function, release of clinic time and overall costs without negatively impacting on patient care.
Understanding the nocebo effect
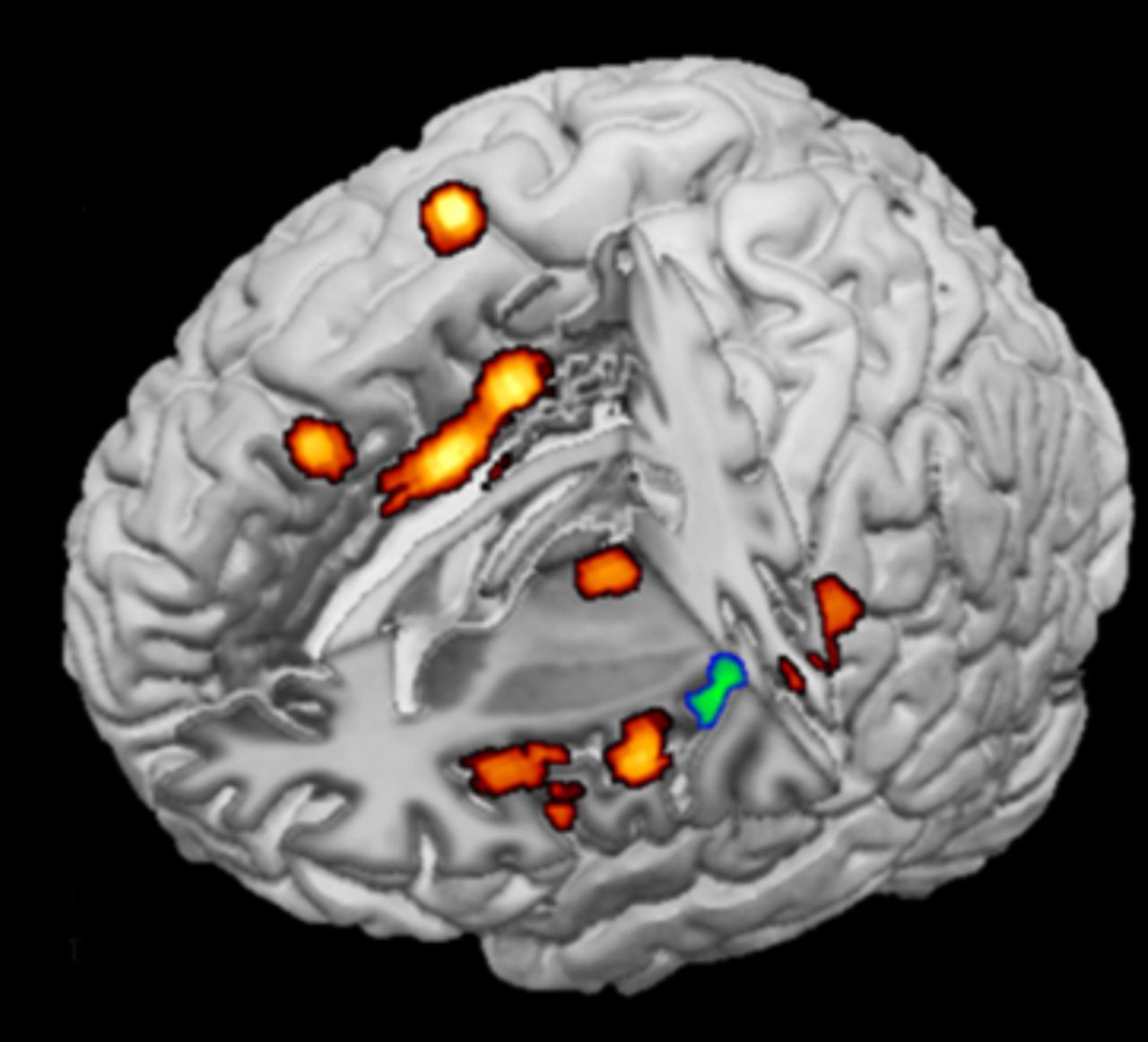
When a patient has the expectation that a given treatment will have no benefit, a nocebo effect, which is the opposite of the placebo effect, can occur in clinical trial and routine care settings.57 If the treatment is given in a negative context, an increase in anticipatory anxiety activates neurochemical systems that increase pain transmission (eg, cholecystokinin and cyclooxygenase), and neuroimaging studies have shown increased activity of brain regions involved in pain processing and emotional regulation (eg, the prefrontal cortex) (figure 5).58–60 When a nocebo response, such as perceived side effects or lack of efficacy, occurs in response to active therapy, it can have a negative impact on treatment adherence and outcomes.59 61 To avoid contributing to the nocebo effect, it is very important that clinicians carefully consider how they communicate with their patients, and make an effort to frame communications in a positive context.59 62

Neuroimaging of a nocebo responder with activations of brain regions involved in anticipatory anxiety projected onto a 3D rendering model of the brain.58 Image courtesy of Fabrizio Benedetti. 3D, three dimensional.
The relevance of the nocebo effect to biosimilars
In a recent survey of 1059 patients with inflammatory bowel disease, 45% of patients were being treated with anti-TNF biological agents, but only 32% of patients had heard of biosimilars.63 Of the patients who had heard of biosimilars (n=383), 47% worried about their safety profile and 39% worried that the biosimilar could be less effective than the reference product, indicating that biosimilars were often viewed in a negative context. Only 26% of patients had no specific concerns about biosimilars.
The potential implications of viewing biosimilars in a negative context are evident in high rates of discontinuation of CT-P13 reported after a switch from reference infliximab in recently published open-label studies.64 65 During 6 months of follow-up in the Biosimilar of Infliximab Options, Strengths and Weaknesses of Infliximab Treatment CHange (BIO-SWITCH) study, 47 of 192 patients (24%) discontinued CT-P13 after a non-mandatory switch because of a perceived lack of effect (n=26), an adverse event (n=11) or a combination of these two reasons (n=10).64 Many of the adverse events that led to withdrawal of CT-P13 were subjective pain-related health complaints, such as arthralgia, headache and myalgia. Furthermore, subjective assessments of disease activity (tender joint count and the patient’s global assessment of disease activity) increased in patients who discontinued CT-P13, but there were no changes in objective assessments of disease activity (swollen joint count or C reactive protein level). Analysis of Danish Registry for Biologic Therapies in Rheumatology (DANBIO) data revealed a discontinuation rate of approximately 15% after 12 months of follow-up for patients switching to CT-P13 from reference infliximab (n=802), as part of a state-mandated switch to biosimilar agents.65 The most frequently reported reason for discontinuation was lack of efficacy, but the switch to CT-P13 did not have a negative impact on disease activity, evaluated 3 months before and after the switch.65 66 Analysis of DANBIO data also showed that 9% of patients who switched from reference etanercept to SB4 (n=1548) discontinued treatment with SB4 during 5 months of follow-up, while disease activity remained largely unchanged 3 months after the switch.66
Communication strategies to avoid nocebo effects
A comparison of the effects of different communication strategies after open-label non-mandatory switching of patients with rheumatic disease from reference infliximab to CT-P13 (BIO-SWITCH study) or from reference etanercept to SB4 (BIOsimilar switch, Study on Persistence and role of Attribution and Nocebo [BIO-SPAN] study) showed that use of an enhanced communication strategy resulted in much higher treatment retention rates (figure 6).67 In both studies, patients received a letter requesting that they switch to a biosimilar, but in the BIO-SPAN study, the request to switch was timed to coincide with a national media feature on biosimilars; lower costs and fewer injection site reactions, as reported in a clinical equivalence trial in patients with rheumatoid arthritis,50 were highlighted to patients as reasons for switching; and healthcare providers received training on how to reduce patient concerns about biosimilars and how to respond to subjective health complaints. Also, whereas patients received CT-P13 infusions in a group setting in BIO-SWITCH, SB4 was administered in individual Subcutaneous (SC) sessions in BIO-SPAN, thereby avoiding a group-think mentality if a patient reverted to the reference product due to perceived lack of efficacy or adverse effects. In general, a shared patient–physician decision to switch to a biosimilar (ie, a non-mandatory switch) using an enhanced communication strategy may help to limit the nocebo effect and be preferable to a mandatory switch.68–70

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Kaplan-Meier curve showing proportions of patients who continued to receive treatment with biosimilar infliximab (CT-P13) in the BIO-SWITCH study or biosimilar etanercept (SB4) in the BIO-SPAN study over 6 months. Shown is the survival curve with 95% CI (shaded areas).67
Conclusions
As the number of anti-TNF bsDMARDs for the treatment of rheumatic diseases increases, significant cost savings will be made as more patients are switched from expensive reference products to more cost-effective biosimilars. However, for bsDMARDs to be widely integrated into clinical practice, and for maximal cost savings to be achieved with these drugs, all rheumatology prescribers and patients need to be aware of the consistent efficacy and safety of bsDMARDs in relation to reference bDMARDs, as well as their substantial cost benefits. Increased awareness of the strict and comprehensive process of evaluation that all biosimilars undergo before and after they are approved is likely to be reassuring and increase successful long-term uptake of biosimilars. Addition of adalimumab biosimilars to the group of anti-TNF bsDMARDs that are already available on the European market will continue to drive savings, allowing rheumatologists to treat a wider range of patients with the most effective available therapies, and for funds to be diverted to other aspects of care, thereby helping to relieve the pressure on tight healthcare budgets. Education and effective communication between healthcare providers and patients are key to dispelling any pre-existing negative perceptions and concerns patients may have about biosimilars, avoiding nocebo effects and facilitating long-term adherence to biosimilars.
Acknowledgments
The authors acknowledge Weber Shandwick Hong Kong for editorial support in the preparation of this manuscript with funding from Samsung Bioepis.
References
Footnotes
Contributors JSS was the lead author of the manuscript. All authors provided critical feedback and commented on the manuscript. All authors approved the final version to be published. All authors agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; internally peer reviewed.
Data availability statement No additional data are available.