Article Text

Abstract
Objective Although clinical trials support equivalence of originator products and biosimilars for etanercept and infliximab, real-world studies among biologics-naïve patients with spondyloarthritis (SpA) are lacking. The objectives were to compare treatment retention in biologics-naïve patients with SpA starting either the originator product or a biosimilar of infliximab and etanercept, and to explore the baseline characteristics of these patients.
Methods Patients with SpA (ankylosing spondylitis/non-radiographical axial SpA/undifferentiated SpA), starting infliximab or etanercept as their first-ever biological disease-modifying antirheumatic drug during January 2014–June 2017 were identified in five Nordic biologics–rheumatology registers. Baseline characteristics were retrieved from each registry; comorbidity data were identified through linkage to national health registers. Country-specific data were pooled, and data on infliximab and etanercept were analysed separately. Comparisons of treatment retention between originators and biosimilars were assessed through survival probability curves, retention rates (2 years for infliximab/1 year for etanercept) and Hazard Ratios (HR).
Results We included 1319 patients starting infliximab (24% originator/76% biosimilar), and 1015 patients starting etanercept (49% originator/51% biosimilar). Baseline characteristics were largely similar for the patients treated with the originators compared with the corresponding biosimilars. Survival probability curves were highly similar for the originator and its biosimilar, as were retention rates: infliximab 2-year retention originator, 44% (95% CI 38% to 50%)/biosimilar, 46% (95% CI: 42% to 51%); and etanercept 1-year retention originator, 66% (95% CI 61% to 70%)/biosimilar, 73% (95% CI 68% to 78%). HRs were not statistically significant.
Conclusion This observational study of biologics-naïve patients with SpA from five Nordic countries showed similar baseline characteristics and very similar retention rates in patients treated with originators versus biosimilars, for both infliximab and etanercept, indicating comparable effectiveness in clinical practice.
- ankylosing spondylitis
- anti-TNF
- epidemiology
- spondyloarthritis
- outcomes research
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Clinical trials support equivalence of originator products and biosimilars for etanercept and infliximab.
Real-world studies among biologics-naïve patients with spondyloarthritis (SpA) are lacking.
What does this study add?
This observational study from five Nordic biologic registries showed comparable patient characteristics and retention to treatment among biologics-naïve patients with SpA treated with originators versus biosimilars (infliximab or etanercept).
How might this impact on clinical practice?
These real-world data indicate similar effectiveness of originator and biosimilar products in SpA.
Introduction
The first infliximab biosimilar (CT-P13) was introduced on the European market in 2014, followed by the first etanercept biosimilar (SB4) in 2015. Before marketing, efficacy and safety of biosimilars versus originators were explored in randomised controlled trials (RCTs). For infliximab, the initial RCTs, PLANETRA1 and PLANETAS,2 were conducted in rheumatoid arthritis (RA) and ankylosing spondylitis (AS), respectively. In the PLANETAS study, the primary outcome was pharmacokinetic equivalence rather than efficacy.2 For etanercept, only patients with RA were studied,3 and so far, no randomised trials have been conducted for SB4 in other indications than RA. However, the regulatory approvals of CT-P13 and SB4 were extrapolated to all of the approved indications for the originator products, thus including the spondyloarthritis (SpA) disease entities.4
Outcomes following a switch from successful treatment with the originator to the corresponding biosimilar drugs (so called non-medical switch) were studied in premarketing extension studies.5–7 Postmarketing, one independent RCT (NOR-SWITCH8) compared safety and effectiveness of infliximab originator (INF) with CT-P13 in routine care. Patients with six different inflammatory disorders (including 121 patients with SpA or psoriatic arthritis (PsA)) were randomised either to continue with INF or to switch to CT-P13, with no differences in outcomes at week 52. Further, several observational studies including patients with SpA have assessed infliximab switch,9–13 in some cases using comparisons with historical cohorts treated with infliximab. Similar studies have also been performed for etanercept.14 The outcome in these observational studies has differed, generally (but not exclusively15) slightly disfavouring the biosimilar product. This has largely been attributed to a nocebo effect, although the evidence for such an effect has been disputed.16 Furthermore, the observed differences could potentially be due to the drugs performing differently in a clinical setting as opposed to the highly selected patients included in RCTs.17
Large observational studies in SpA comparing biologics-naïve patients starting biological disease-modifying antirheumatic drug (bDMARD) treatment with an originator versus treatment with their respective biosimilar during the same calendar time period are thus lacking. Such studies are of particular importance since the only study leading to registration that included patients with AS (PLANETAS2) did not have efficacy as its primary outcome.
The market entry of the biosimilars has been accompanied by a significant reduction in costs in many countries, which has spurred a transition from the originator products towards their biosimilars. In the Nordic region (Denmark, Iceland, Finland, Norway and Sweden), the strategies for transition have differed markedly across the countries, depending primarily on differences in pricing.18 Within Sweden, the uptake was further diversified by separate tender processes in different counties.19 As a result, during the period 2014–2017, biologics-naïve patients with SpA in the Nordic countries have started both originator and biosimilar versions of infliximab and of etanercept. This offers a unique opportunity to compare the performance of these drugs in biologics-naïve patients with minimal risk of a nocebo effect.
The main objective of this study was therefore to compare the treatment retention for infliximab and etanercept originator (INF and ETN) products with their respective biosimilars (CT-P13 and SB4), in bio-naïve patients with SpA starting treatment in 2014–2017, and furthermore to explore the baseline characteristics of treated patients toassess comparability.
Patients and methods
Study design
This is an observational cohort study.
Biologics in the Nordic countries
The strategies for implementing biosimilars in routine care have varied between Nordic countries. Thus, the countries have applied different policies towards switching and choice of biosimilars/originators in biologics-naïve patients. In Denmark, it was mandatory to switch all patients treated with INF or ETN to their respective biosimilars, when the biosimilars were marketed.9 14 20 In Sweden, the strategies for both new starts and switching differed across the different counties,19 and in Finland, INF or a biosimilar was used based on exclusive pricing negotiations made in individual hospital districts, whereas the first etanercept biosimilar was not reimbursed in Finland until January 2018. In Iceland, the least expensive bDMARDs are recommended as first-line drugs based on an annual tendering process under the authority of the Icelandic Health Insurance (state insurance company). Switching from INF to its biosimilars is also recommended when the biosimilar is marketed in Iceland (biosimilar to etanercept was not marketed in Iceland at the study time). In Norway, non-medical switching policies differ between hospitals, whereas the decision between biosimilars/originators in biologics-naïve patients is decided by the annual national tender.
Case definitions
Patients with SpA starting infliximab or etanercept (originator or biosimilar) as their first bDMARD at any time between 1 January 2014 and 30 June 2017 were identified from the national biologics registers in Sweden (ARTIS), Denmark (DANBIO), Finland (ROB-FIN), Iceland (ICEBIO), as well as a regional biologics register in Norway (NOR-DMARD). In all these registries, information on patient characteristics, disease activity and treatment with DMARDs is registered prospectively as part of routine care as previously described.18
The registers define the SpA diagnoses differently, either based on International Classification of Diseases, 10th Revision (ICD-10) codes or on clinical diagnoses. SpA was therefore defined as having either an ICD-10 code for AS (M45) or undifferentiated SpA (M46.0, M46.1, M46.8 and M46.9), or a clinical diagnosis of AS or non-radiographical axial SpA. Patients with a primary diagnosis of PsA were excluded, but an additional diagnosis of cutaneous psoriasis (identified from the national patient registries; see description below) was allowed.
Patient characteristics
Patient characteristics at baseline (ie, when starting the first bDMARD treatment) were identified in the biologic registries according to the visit closest (within −30 days to +14 days) to the start date of the bDMARD in question. Characteristics included SpA disease duration, patient pain score on a visual analogue scale (VAS) (0–100 mm), patient global score on a VAS (0–100 mm), C reactive protein (CRP) level (mg/L), Ankylosing Spondylitis Disease Activity Score (ASDAS), Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) and Bath Ankylosing Spondylitis Functional Index (BASFI). Furthermore, concomitant use of a conventional synthetic disease-modifying antirheumatic drug (csDMARD) (yes/no) was registered.
Linkage to a National Patient Register was available in Denmark21 and Sweden,22 from which data were retrieved on inpatient and outpatient contacts for diagnoses of the extra-articular SpA manifestations of psoriasis (ICD-10: L40) and inflammatory bowel disease (ICD-10: K50, K51) in the last 10 years prior to baseline. In Finland, a similar linkage was made to the Care Register (HILMO) provided by the National Institute for Health and Welfare (THL).
Treatment retention
Start of follow-up was defined as the start date of the first bDMARD. End of follow-up was defined as the date of treatment discontinuation (for those who did discontinue), or on censoring defined as the first of death or the end of the study period (30 June 2017). For patients who switched from the originator/biosimilar to its biosimilar/originator, follow-up was censored at the switch date.
Treatment response
Patient global and patient pain score, BASDAI, ASDAS, CRP and BASFI were assessed at baseline and at 6 months after the start of follow-up, defined as the visit closest to 180 days after treatment start (within days 90–270).
Statistics
Baseline characteristics are presented as means or proportions. For each of the comparative analyses described further below, available data from the five Nordic countries were pooled.
Treatment retention was compared between the originator and biosimilar products through crude survival probability curves, truncated at 1 year of follow-up for etanercept and at 2 years for infliximab, with a crude statistical comparison through log-rank test. For etanercept, the assessment of treatment retention was limited to 1 year since SB4 was introduced 1 year after CT-P13, resulting in few patients remaining at risk and still treated after 1 year. Retention rates were calculated at 1 year for etanercept and at 1 and 2 years for infliximab.
HRs for discontinuing treatment were estimated using Cox proportional hazard models: crude and in three models adjusting for baseline values of (1) age, sex and concomitant csDMARD (yes/no/missing); (2) age, sex, csDMARD and CRP (categorised in quartiles); and (3) age, sex, csDMARD and patient global score (categorised in quartiles). Missing values due to missing information or a missing visit at baseline were added as a separate category. Adjusting for CRP and patient global score rather than the disease-specific activity measures ASDAS or BASDAI was chosen due to a higher proportion of missing data for the latter. No evidence of departure from the proportionality assumption was observed in these models.
The proportions of patients contributing data at baseline and at 6 months of follow-up are presented in online supplementary table S1. Due to a high proportion of missing disease activity measures, no statistical comparisons were made; only group means and SDs are presented.
Supplemental material
Patient and public involvement
Within the Nordic collaboration performing this study, patient representatives were involved in the planning and development of the research questions and the study design.
Results
In total, 2334 patients with SpA were included, of whom 1319 started infliximab and 1015 started etanercept as their first biological treatment. There were no clinically relevant differences in baseline characteristics and demographics between the originator and corresponding biosimilar cohorts, apart from a higher proportion of patients using concomitant csDMARD in the originator cohorts of both infliximab and etanercept (table 1). The use of biosimilars versus originators differed markedly between the countries. In Finland and Iceland, SB4 was not available during the time period.
Baseline characteristics and demographics of infliximab-treated and etanercept-treated patients with SpA and reason for discontinuation
Treatment retention
The survival probability curves for the first 2 years of treatment were similar for CT-P13 and INF (p value of 0.59), and so were 1 year probability curves for ETN and SB4 (p value of 0.18) (figure 1A,B).
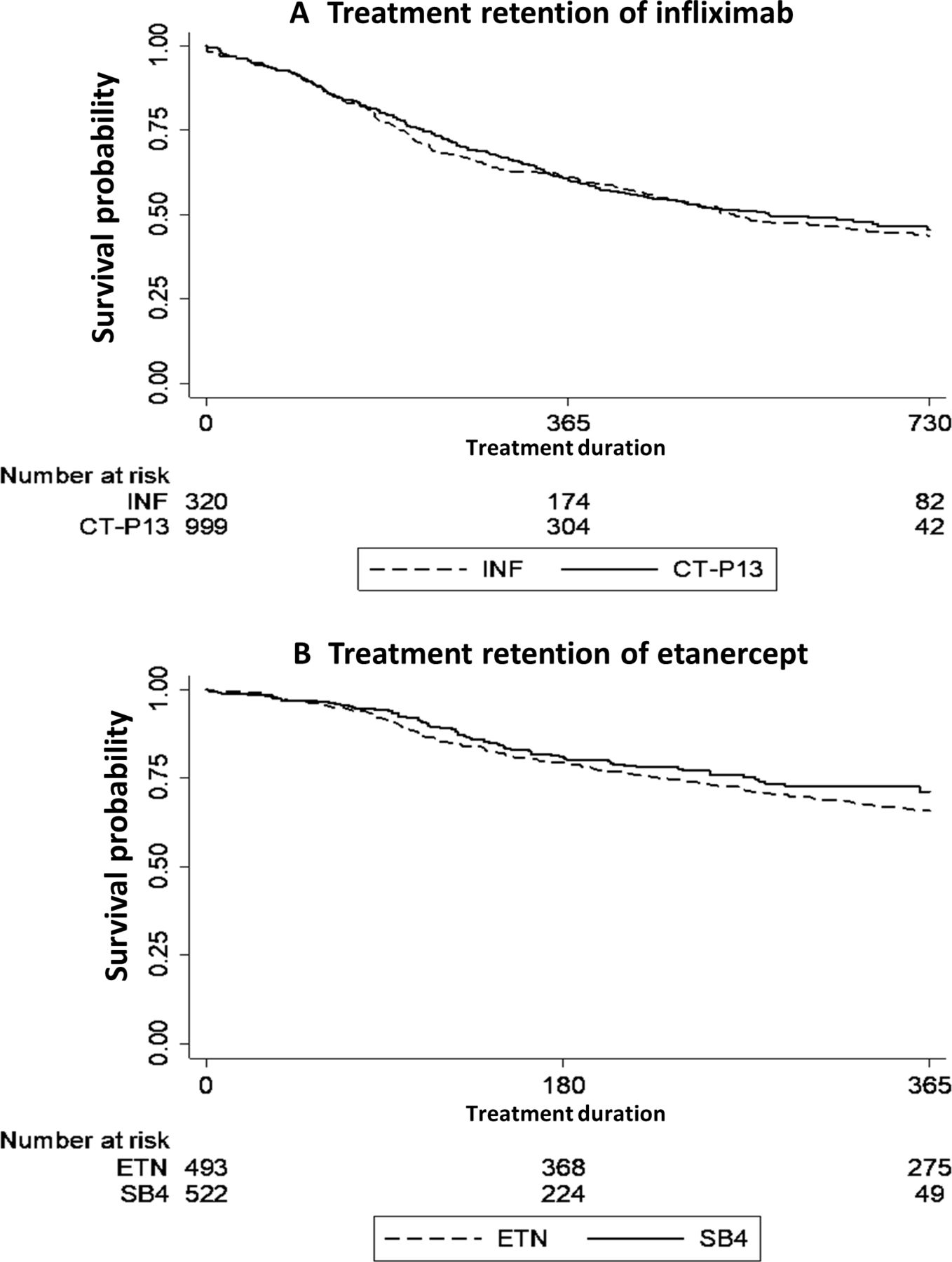
{kind=link}
Treatment retention in patients treated with infliximab or etanercept biosimilar and originator products. Kaplan-Meier curves of treatment retention for (A) CT-P13 and INF, and (B) SB4 and ETN. CT-P10, infliximab biosimilar; ETN, etanercept originator; INF, infliximab originator; SB4, etanercept biosimilar.
The 1-year retention rates for INF and CT-P13 were comparable: 1-year INF, 62% (95% CI 57% to 68%); CT-P13, 63% (95% CI 60% to 66%); 2-year INF, 44% (95% CI 38% to 50%), and CT-P13, 46% (95% CI: 42% to 51%). Likewise, the 1-year retention rates for ETN and SB4 were comparable: ETN, 66% (95% CI 61% to 70%) and SB4, 73% (95% CI 68% to 78%).
The proportional hazard analyses showed no statistically significant difference in treatment retention when comparing the biosimilars with their respective originator products, neither in crude nor in adjusted analyses (table 2). The results indicated a 5% decrease in risk of discontinuation favouring CT-P13 compared with INF, and a 15% decrease for SB4 compared with ETN; however, in neither case did the 95% CIs confirm a statistically significant decrease in risk. Adjusting for baseline use of concomitant csDMARD, which did differ between the originators and biosimilars, did not change the results.
HRs for discontinuing treatment with (A) biosimilar versus originator (reference) infliximab, (B) biosimilar versus originator (reference) etanercept
Disease activity at baseline and 6 months
Table 3 presents mean values (SD) for patient global and pain scores, BASDAI, BASFI, CRP and ASDAS at baseline and at 6 months of follow-up. At both baseline and 6 months of follow-up, mean values showed similar disease activity for the two biosimilars compared with their originators.
Mean disease activity at baseline and after 6 months of follow-up for patients treated with infliximab or etanercept
Discussion
In this observational study of 2334 biologics-naïve patients with SpA initiating treatment with infliximab or etanercept, we found highly similar treatment retentions for the originator products compared with their biosimilars. The characteristics of the patients receiving the originators and the biosimilars were also similar, suggesting that the cohorts were comparable. Thus, these results support the equivalence of the treatments.
Due to the complex manufacturing process, a biosimilar product is never 100% identical to its originator.23 Thus, although equivalent efficacy and safety of the biosimilar must be demonstrated before marketing, it has been debated how to implement biosimilars in routine care.24 Furthermore, concerns have been raised regarding extrapolation across indications.
Several studies have assessed the effect of switch to a biosimilar, performed among patients treated with the originators, some including patients with SpA. The first of these were the extension studies of the initial RCTs5–7 and the RCT study NOR-SWITCH8 (now with an extension25), followed by a small observational study,10 none of which found differences in effectiveness following a switch from INF to CT-P13 or ETN to SB4. However, in observational studies investigating the effect of non-medical switching, it is challenging to find a comparable control group, since only an RCT design (such as NOR-SWITCH) can eliminate channelling (towards treatment) completely. In one large observational study assessing infliximab switch, a comparison was performed with a historical INF cohort, and a slightly lower retention rate was described for the patients switched to CT-P13.9 Similar results were found in a large observational study of ETN to SB4 switch.14 In another study, 89 patients (75 with SpA) switching from INF to CT-P13 were compared with 29 infliximab-naïve patients starting CT-P13 and 82 historical patients starting INF, also finding a lower retention rate for the switched patients compared with the other two groups.12 Yet another small study evaluating the INF to CT-P13 switch found a discontinuation rate of 24% during 6 months of follow-up after switching.11 In both of these latter studies,11 12 the increased discontinuation rate for the biosimilar appeared to be driven by a subjective patient perception of disease activity rather than objective measures, supporting a nocebo effect. Yet another study, assessing the infliximab switch, estimated a ‘nocebo response’ of 13%, further suggesting that a shared decision-making and patient involvement in the switch process may lead to favourable retention rates.13 The observational studies assessing the effect of switching, either comparing with non-switched patients or comparing with a historical cohort, are thus hampered by potential confounders, such as patients’ expectations towards the switch (nocebo), which cannot easily be disentangled from actual differences in the effect, or confounding by indication/channelling towards the choice of drug. Hence, such studies do not necessarily reflect the true biosimilar drug effect.
A strength of the present study is that confounders, such as nocebo related to the context of switching from a stable ongoing treatment with an originator to its (cheaper) biosimilar, should be minimised in a bionaïve population. Another strength is that the pooling of data from the different countries ought to reduce the risk of country-specific channelling towards originators or biosimilars.
Our study also has some limitations. First, the pooled data from the five different Nordic countries did not allow for a direct comparison of effectiveness (eg, response rates) of the originators and the biosimilars due to missing data. However, the similarities in the mean disease activity measures at baseline and at 6 months of follow-up, together with the very similar drug retention, suggest similar effectiveness. Another potential problem is that not all countries contributed to all treatment arms. We have previously explored treatment of SpA with bDMARDs in the Nordic countries and demonstrated that the most notable difference is in the use of concomitant csDMARDs, which we have adjusted for in the analyses.18
In conclusion, in this large, international, biologic-naïve SpA cohort, we found equivalent treatment retentions for infliximab and etanercept biosimilars compared with their originators, indicating comparable effectiveness in routine care.
Acknowledgments
We thank all the departments contributing to the clinical data collection in the participating biologic registers, as well as the participating patient representatives. Thank you to Frank Mehnert, Clinical Epidemiology, Aarhus University Hospital, Aarhus, Denmark, who contributed management of data from the Danish National Patient Registry.
References
Footnotes
UL and BG are joint first authors.
Contributors UL, BG and LJ contributed to the study design. DDG performed the analysis of raw data. All authors contributed to the interpretation of the results and in the preparation of the manuscript.
Funding This study was partly funded by grants from Nord-Forsk and FOREUM.
Competing interests BG: grant/research support from Biogen, Pfizer and AbbVie. JA: Karolinska Institutet (through JA) has or has had research agreements with the following pharmaceutical companies, mainly in the context of the ARTIS national safety monitoring programme for rheumatology biologicals: Abbvie, BMS, MSD, Eli Lilly, Pfizer, Roche, Samsung Bioepis and UCB; Karolinska Institutet has alo received remuneration for JA participating in ad boards arranged by Lilly, Novartis and Pfizer. DN: grant/research support from MSD, Pfizer; consultant for AbbVie, BMS, MSD, Novartis, Roche, Pfizer and UCB; speakers' bureau: Novartis and UCB. SP: consultant for Novartis; speakers' bureau: Lilly. MH: grant/research support from BMS, MSD, AbbVie, Roche, Novartis, Biogen and Pfizer; consultant for Eli Lilly; speakers' bureau Orion Pharma, Biogen, Pfizer, CellTrion, Merck and Samsung Bioepis. LJ: lecture and consulting fees from Pfizer, Abbvie, Novartis, Eli-Lily and Janssen.
Patient consent for publication Not required.
Ethics approval The appropriate ethical committees and/or data protection committees in each country approved of the study (approval codes for Sweden: 2015/1844-31/2; Denmark: RH-2015–209, I-suite 04145; Norway: 2011/1339 and 2017/243; Finland: 73/13/03/00/2014; and Iceland: VSNb2017010049/03.01). Individual patient consent was not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.