Article Text

Abstract
Background/Objective FKB327 is a biosimilar of the antitumour necrosis factor adalimumab reference product (RP). A randomised, double-blind (DB) phase 3 study compared the efficacy of FKB327 with the RP in patients with active rheumatoid arthritis (RA) inadequately controlled with methotrexate (MTX). A subsequent randomised open-label extension (OLE) study with treatment switching assessed long-term safety, efficacy, pharmacokinetics and immunogenicity of FKB327 compared with the RP.
Methods Patients with moderate-to-severe, active RA on a stable dose of MTX were randomised 1:1 to receive FKB327 or the RP (40 mg subcutaneously every other week) for 24 weeks. Patients who completed the DB study were enrolled in the OLE and rerandomised 2:1 to receive FKB327 or the RP; two-thirds continued on the same treatment and one-third switched for 30 weeks. All patients received FKB327 through Week 76. Long-term efficacy, safety and immunogenicity were assessed.
Results Of 728 patients in the DB study, 645 were enrolled in the FKB327-OLE study. The American College of Rheumatology (ACR)20 response rates for all treatment groups at Week 30 in the OLE ranged from 83.2% to 85.9%. ACR20 response rates remained stable for all patients regardless of single- or double-switching treatment and were similar for all treatment sequences through Week 76. The safety profile and incidence of antidrug antibodies were comparable across sequences.
Conclusion Efficacy, safety and immunogenicity were similar among patients with RA treated with FKB327 or the RP for up to 2 years, and were not affected by single- or double-switching treatment.
- rheumatoid arthritis
- treatment
- anti-TNF
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
INTRODUCTION
Biologic disease-modifying antirheumatic drugs (DMARDs) have been a major advance in the treatment of patients with rheumatoid arthritis (RA).1 2 Adalimumab, a recombinant human monoclonal antibody against tumour necrosis factor (TNF)-alpha, was initially approved in 2002 in the United States and in 2003 in the European Union for the treatment of RA. In addition, adalimumab is indicated for the treatment of patients with juvenile idiopathic arthritis; psoriatic arthritis; ankylosing spondylitis/axial spondyloarthritis; hidradenitis suppurativa; plaque psoriasis and Crohn’s disease; adult and pediatric ulcerative colitis; and noninfectious intermediate, posterior and panuveitis in adult patients.3 4 FKB327 was developed as a biosimilar of the adalimumab reference product (RP).
Key messages
What is already known about this subject?
Adalimumab is a tumour necrosis factor inhibitor that is effective in treating patients with moderate-to-severe rheumatoid arthritis and other chronic immune-mediated inflammatory conditions.
What does this study add?
FKB327 is a biosimilar to the adalimumab reference product (RP) and demonstrates similar efficacy, safety and immunogenicity compared with the RP in long-term studies.
The biosimilarity in efficacy, safety and immunogenicity was not affected by switching or double-switching treatment between the adalimumab RP and FKB327.
How might this impact clinical practice or future developments?
These data will help inform clinician decision-making regarding switching from the adalimumab RP to FKB327 and may result in increased patient access to biological therapies.
Adalimumab is administered at a dose of 40 mg/0.8 mL or 40 mg/0.4 mL in a single-use prefilled syringe or pen every other week (EOW) via subcutaneous injection for adult patients with RA; FKB327 was delivered at the same dose, in the same manner.3 FKB327 is a biosimilar to the adalimumab RP that contains the same active ingredient but different excipients, including monosodium glutamate, sorbitol, methionine, polysorbate 80, hydrochloric acid (for pH adjustment) and water for injections, and excludes sodium citrate.
FKB327 has demonstrated a similar pharmacokinetic (PK) profile in healthy subjects with a single subcutaneous dose.5 Data regarding switching from the RP to biosimilars in addition to long-term treatment are desirable to strengthen the demonstration of biosimilarity and reassure prescribers and users regarding the safety of switching. No increased risk in safety and immunogenicity has been observed in 1-year treatment with other adalimumab biosimilars.6–8 However, further evidence with long-term treatment, including treatment switching, is needed in treatment with TNF-alpha inhibitors in chronic inflammatory diseases.
The primary objective of this double-blind (DB) study and open-label extension (OLE) was to evaluate the safety and efficacy of treatment with FKB327 compared with the RP when each was administered in combination with methotrexate (MTX) in patients with RA. Preliminary data through 54 weeks of treatment have been published previously.9 The current data set evaluated the long-term efficacy and safety of the combination of FKB327 plus MTX compared with the RP plus MTX for up to 2 years of treatment. The current study was also designed to investigate the long-term effects of single-swiching treatment and to assess any effects of double-switching treatment for the first time in this treatment population.
METHODS
Study design
The study design of the DB study (Period 1) and the first 30 weeks of the OLE (Period 2) has been described in greater detail by Genovese and colleagues.9 Briefly, the DB study was a phase 3, randomised, parallel-arm, active comparator–controlled, 24-week equivalence study (NCT02260791) designed to evaluate the similarity in the efficacy of FKB327 to the RP and compare PK, safety and immunogenicity in patients with RA inadequately controlled with MTX.
In the DB study, 730 patients were randomised in a 1:1 ratio to receive either FKB327 40 mg or the RP 40 mg administered as a subcutaneous injection EOW, stratified by an interactive web response system using dynamic randomisation method, by prior biological treatment and Disease Activity Score in 28 joints (DAS28) using C-reactive protein (CRP) ≤5.1/>5.1. FKB327 was provided in vials, whereas the RP was supplied in a prefilled syringe. To ensure blinding of patients and study staff, a staff member (who was not otherwise involved in the study) filled syringes and placed them into a masking unit to allow the dose to be administered without revealing the syringe appearance. In addition, a nurse administered injections without allowing patients to see the syringe. Clinic visits were EOW, starting at Week 0 and extending through Week 22 (Period 1).
The subsequent randomised OLE with treatment switching (NCT02405780) assessed long-term safety, efficacy, PK and immunogenicity. Patients who received FKB327 in the DB study were rerandomised to receive FKB327 or the RP in a 2:1 ratio so that two-thirds remained on the same treatment as in the FKB327-002 study and received the study drug with the same dosage regimen for 30 weeks in Period 2, whereas one-third were switched to the alternative treatment (figure 1).
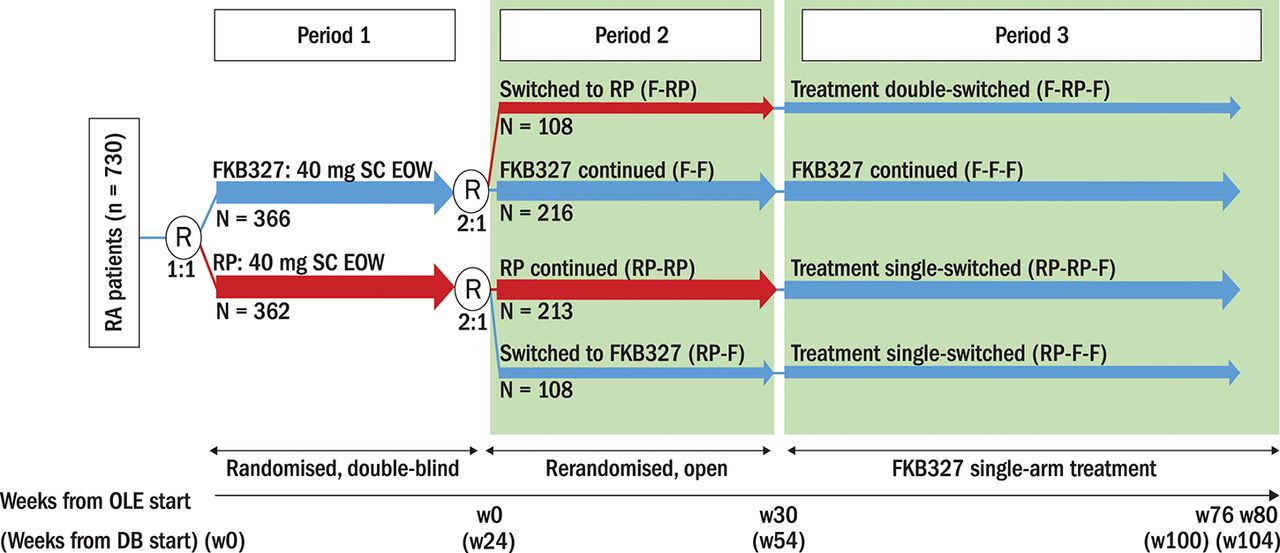
Study schema. DB, double-blind; EOW, every other week; F, FKB327; OLE, open-label extension; R, randomisation; RA, rheumatoid arthritis; RP, reference product; SC, subcutaneous; w, week. *All patients (except US) were introduced to the FKB327 auto-injector during Period 3.
All patients received FKB327 until Week 76, followed by a 4-week follow-up period (Week 80) in Period 3, which is the long-term extension of the previously described primary analysis.9 In Period 3, 100 patients experienced a double switch (FKB327 [F]-RP-F) and 190 experienced a single switch (RP-RP-F), whereas the remaining patients continued FKB327 treatment (figures 1 and 2). Therefore, patients received FKB327 and/or the RP for a total of 100 weeks (last observation, Week 104) from the start of the DB study. Clinic visits were every 4 to 12 weeks in Period 2 and every 12 weeks in Period 3. FKB327 (40 mg/0.8 mL) was administered in a prefilled plastic syringe with a safety device for single use only in the OLE study, and US-licensed Humira (adalimumab; 40 mg/0.8 mL) was supplied as the RP in the study in Period 2 and was administered in a prefilled Type 1 glass syringe. In the FKB327 single-arm treatment phase in Period 3, the FKB327 auto-injector (40 mg/0.8 mL) was introduced, except to US patients, due to regulatory considerations. Patients in the OLE were enrolled from 92 sites in 11 countries.
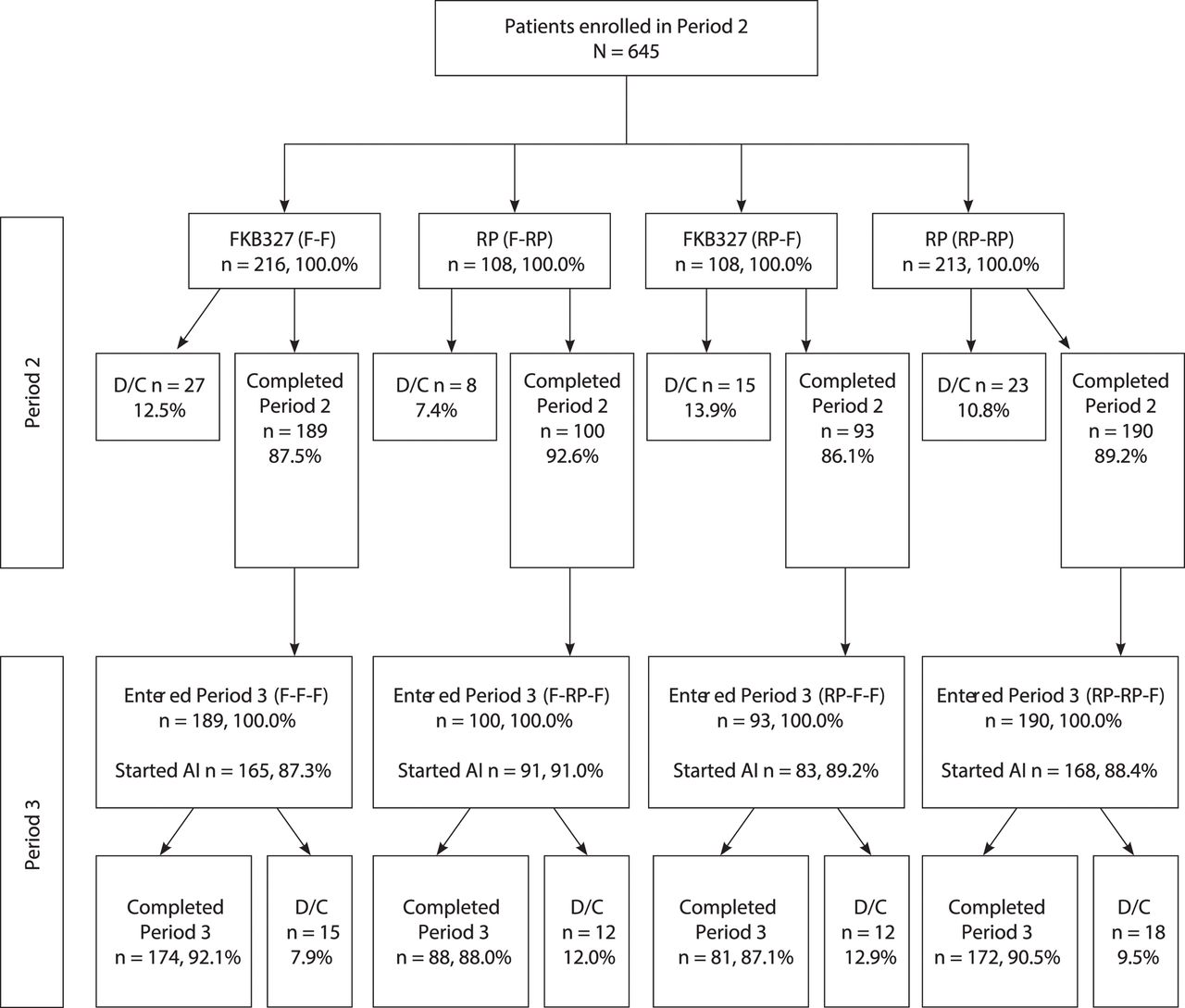
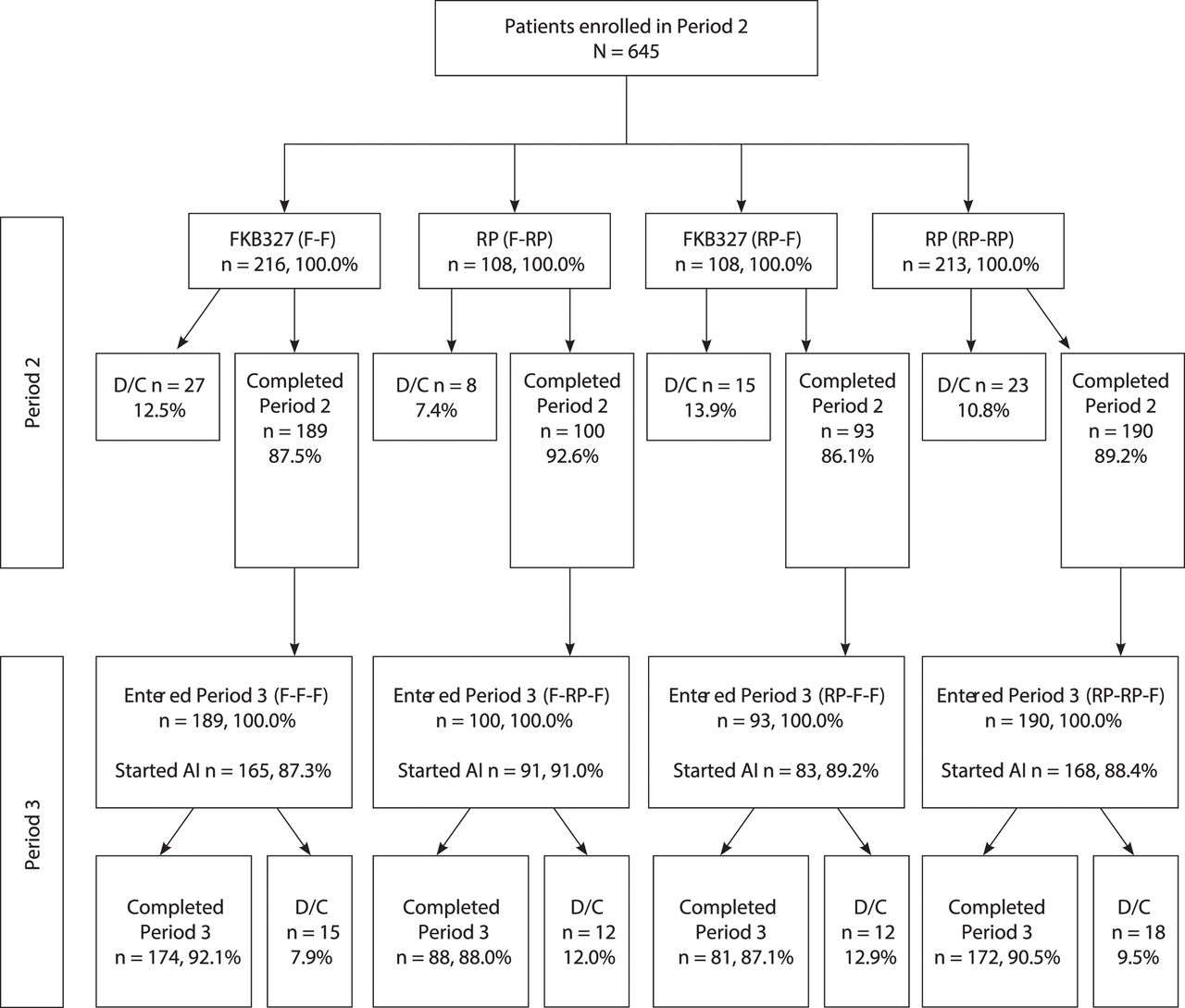
Consort diagram. D/C indicates prematurely discontinued; DB, double-blind; F, FKB327; RP, reference product. Note: All patients (except US) were introduced to the FKB327 auto-injector during Period 3.
The study was conducted in accordance with the Declaration of Helsinki and International Conference on Harmonisation Guidelines for Good Clinical Practice. Study protocols were reviewed and approved by an independent ethics committee or institutional review board for each study centre. Written informed consent was obtained from all patients before entry in the DB and OLE studies.
Patients
Inclusion and exclusion criteria were described in greater detail by Genovese and colleagues.9 Briefly, enrollees were aged ≥18 years with moderate-to-severe, inadequately controlled RA despite ≥3 months of treatment with MTX; ≥6 tender joint count (TJC) and swollen joint count (SJC) at screening and baseline; and CRP ≥10 mg/L at screening. Patients were ineligible if they had prior treatment with adalimumab, ≥1 biologics or 1 DMARD, or intra-articular or parenteral steroids within 28 days of screening; clinically significant laboratory abnormalities; cardiovascular disease; demyelinating diseases; chronic or acute infection; body weight >120 kg; or positive QuantiFERON Gold blood test for latent tuberculosis. Patients received MTX (10 mg-25 mg/week stable dose) during the study.
At Week 24, at the investigators’ discretion, patients who completed the DB study with clinical response and no serious adverse events (AEs) were eligible to enter the OLE and undergo rerandomisation in a 2:1 ratio, so that two-thirds of patients remained on the same treatment and one-third switched to the alternate treatment (40 mg subcutaneously EOW) for Weeks 0 through 28 (Period 2). Subsequently, all patients received FKB327 from Week 30 through Week 76, followed by a 4-week follow-up period (Period 3).
Therefore, the four patient groups based on treatment in the DB study and in the two periods of the OLE (figure 2) included
Patients who received FKB327 during the DB study (Period 1) and were rerandomised to the same treatment in the OLE (Periods 2 and 3) (F-F-F)
Patients who received the RP during the DB study (Period 1) and were rerandomised to the same treatment in Period 2 (RP-RP-F)
Patients who received FKB327 during the DB study (Period 1) and were rerandomised to the RP in Period 2 and switched back to FKB327 in Period 3 (F-RP-F)
Patients who received the RP during the DB study (Period 1) and were rerandomised to FKB327 in the OLE (Periods 2 and 3) (RP-F-F).
Study end points and statistical methods
DB study
In the DB study, the primary efficacy end point was the American College of Rheumatology (ACR)20 response rate at Week 24. Biosimilarity was determined according to the recommendations of the EU Committee for Medicinal Products for Human Use (CHMP) and the US Food and Drug Administration (FDA). The percentage of patients achieving ACR20, ACR50 and ACR70 response, and DAS28-CRP were secondary end points during the DB study. In addition, ACR20/50/70 response rates and changes in DAS28-CRP scores from baseline of the DB study were analysed by treatment sequence in the OLE study.
Open-label extension
In the OLE, the primary end point was safety. Safety through Period 2 has been described previously in a preliminary analysis.9 The current final analysis evaluates safety across both Period 2 and Period 3 for patients who underwent single and double switching between FKB327 and the RP. Safety was monitored throughout the study by evaluating the incidence of treatment-emergent AEs (TEAEs) and AEs of special interest (eg, infections). The number of patients experiencing TEAEs by each period and the exposure-adjusted number of events (number of events divided by patient-year) was determined. Efficacy end points included ACR20/50/70 response rates, and DAS28-CRP at each clinic visit. PK was assessed by evaluation of serum concentrations of adalimumab, using a validated immunoassay on an electrochemiluminescent platform with the lower limit of 100 ng/mL1.5 Immunogenicity was assessed by evaluation of antidrug antibodies (ADAs) by single-assay approach. Sensitive electrochemiluminescence bridging format (Meso Scale Discovery) with acid dissociation was used for the ADA assay to increase drug tolerance in the repeated dosing study.10 Neutralising ADAs were assessed by sensitive competitive ligand binding.11 ADA titres were assessed, and results were summarised using the following imputed categories: 0.0625, 0.25, 1, 4, 16, 64, 256, 1024, 4096, 16 384 and 65 536. The frequency of ADA-neutralising results was summarised for each treatment sequence for the overall treatment period, and for each treatment group for each period by time point.
The safety population comprised all patients in the OLE who received ≥1 treatment doses in the DB study. Efficacy analysis was based on the Full Analysis Set, defined as the set of patients who received ≥1 doses of the randomised treatment with ≥1 evaluable efficacy measurements. Patients were analysed according to their randomised treatment sequence across both studies or randomised treatment group to ensure statistical testing was not biased by a nonrandom assignment. The mean percentage of patients who achieved an ACR20 response and DAS28-CRP score at each postbaseline time point from the DB study was calculated by treatment sequence across the OLE, using 95% confidence intervals (CIs) via the Clopper-Pearson method. Missing responses for the ACR measurements and responses for patients who discontinued treatment before Week 24 were imputed as follows: if the patient withdrew due to lack of efficacy, withdrawal of consent, an AE (non-infectious) or a medical reason (non-infectious), or if the patient had taken a prohibited treatment for RA and had been withdrawn from study treatment, the patient was regarded as a ‘nonresponder’; for all other patients with a missing ACR response at Week 24, the last observation carried forward was used to determine whether they were ‘responders’ or ‘non-responders.’ Efficacy end points were analysed by four treatment sequences across the studies. All analysis data sets and output were produced by the Biostatistics Department of Quanticate UK Limited, using the SAS system Version 9.3 (Unicode Support).
RESULTS
Patients
DB study
In total, 1327 patients were screened; 597 patients failed screening. Subsequently, 730 patients were randomised to FKB327 (n=367) and the RP (n=363). Of these, 366 patients (99.7%) were randomised to FKB327 and 362 patients (99.7%) were randomised to the RP and received the study drug (figure 1). The treatment groups were well balanced with respect to demographic and baseline disease characteristics (table 1).
Summary of demographics and baseline characteristics: safety analysis set
The number of patients treated each week was similar in the FKB327 and RP treatment groups. Overall, 333 patients (90.7%) in the FKB327 group and 328 patients (90.4%) in the RP group completed the study. Of these, 16 patients (FKB327 group, 9 [2.7%]; RP group, 7 [2.1%]) did not proceed to the OLE study due to patient preference or investigator opinion; all other patients continued to the OLE study.
Open-label extension
In total, 645 eligible patients who completed the DB study and consented to the OLE (88.4%) continued into the OLE and were rerandomised to treatment in Period 2 (as shown in figure 2 and previously published by Genovese and colleagues).9 Of the 324 patients who received FKB327 in the DB study, 216 patients were rerandomised to FKB327 (F-F) and 108 were switched to the RP (F-RP) in Period 2. Of the 321 patients who received the RP in Period 1, 108 patients were rerandomised to FKB327 (RP-F) and 213 were rerandomised to the RP (RP-RP) in Period 2. In Period 3, all patients received FKB327, resulting in treatment groups of F-F-F (no switch), F-RP-F (double switch), RP-F-F (single switch) and RP-RP-F (single switch), which represent longer-term results and the effect of double switching.
Most patients (82.0%) had not received biological treatment before the DB portion of the study. Mean DAS28-CRP at baseline in the OLE was 3.5 and 3.4 in the FKB327 and RP treatment groups, respectively. A total of 572 patients had completed 28 weeks of randomised study treatment in Period 2, of which 189 patients had received continuous treatment with FKB327 and 190 patients with the RP (table 2). The OLE treatment groups and the overall treatment sequences were well matched with respect to demographic characteristics in Period 2,9 with the exception of a higher proportion of elderly patients receiving the RP (18.7%) compared with FKB327 (13.9%).
Summary of patient disposition: all enrolled patients
Safety
The incidence of TEAEs was numerically lower in patients treated with FKB327 compared with patients treated with the RP (1.707 vs 2.075 events per patient-year, respectively). However, the incidence of TEAEs leading to premature discontinuation was numerically higher for FKB327 than for the RP (0.091 vs 0.063 events per patient-year, respectively), possibly resulting from longer overall study treatment duration for FKB327. The incidence of TEAEs leading to temporary interruption of study treatment was numerically lower in patients treated with FKB327 than with the RP (0.129 vs 0.171 events per patient-year). The incidence of treatment-emergent serious AEs (TESAEs) leading to temporary interruption of study treatment was similar for the treatments (0.024 and 0.023 events per patient-year, respectively). The incidence of deaths was the same for the two treatments (0.006 events per patient-year for FKB327 and the RP). Although the incidence of TESAEs for FKB327 and RP was the same (0.091 events per patient-year), the incidence of TESAEs leading to discontinuation was numerically higher for FKB327 than for the RP (0.025 vs 0.011 events per patient-year).
The most common treatment-related TEAEs (reported for ≥1% of patients overall) were summarised in the Supplementary Table. Overall, 208 patients (32.2%) experienced a TEAE considered related to the study drug by the investigator, and of those, the most common treatment-related TEAEs were bronchitis, nasopharyngitis and urinary tract infection [UTI]). The most frequently reported TESAEs were infections and infestations, in 10 patients receiving FKB327 (1.6%) and 5 patients receiving the RP (1.6%). Serious infections with the RP included single cases of pneumonia, acute pyelonephritis, bronchitis, appendicitis and pulmonary mycosis. For FKB327, six patients experienced serious AEs of pyelonephritis (including acute cases), four patients experienced pneumonia and two patients experienced sepsis. The safety data for Period 3 were generally consistent with those observed during Periods 1 and 2.
Supplemental material
A numerically lower overall incidence of infections was observed in patients treated with FKB327 than with the RP (0.557 vs 0.684 events per patient-year), but the incidence of serious infections was comparable (0.024 vs 0.029 events per patient-year; table 3). A numerically lower incidence of injection-site reactions (ISRs) and events suggesting hypersensitivity reaction or anaphylaxis to study drug was observed for FKB327 compared with the RP. Six patients receiving FKB327 experienced eight events of neutropaenia (incidence rate [IR] of 0.012 events per patient-year), compared with one patient with one event on the RP (IR of 0.006 events per patient-year). Three patients (0.5%) receiving FKB327 had a malignancy (0.004 events per patient-year; one case [RP-F-F] in Period 2, two cases [RP-RP-F; F-F-F] in Period 3) compared with none on the RP, although two of three patients had received the RP previously. No patient developed pancytopaenia/aplastic anaemia, thrombocytopaenia, a demyelination event or a lupus-like reaction, all known risks with adalimumab.
Summary of treatment-emergent adverse events of interest: safety analysis set
A secondary objective of this study was to evaluate safety for patients switched from FKB327 in the preceding DB study to the RP in the OLE, and then switched back to FKB327 in the third period (from Week 30; double switch; ie, patients in F-RP-F [N=100]). During Period 3, 340 patients (59.4%) experienced ≥1 TEAEs. This was numerically higher than the 52.4% observed during Period 2, when patients were receiving FKB327 and the RP. Incidences were similar for the F-RP-F (61.0%), F-F-F (60.3%) and RP-RP-F (60.0%) sequences and were numerically lower in the RP-F-F sequence (54.8%). Similar to Period 2, the most commonly reported TEAEs were nasopharyngitis, UTI, bronchitis and upper respiratory tract infection.
ACR20 response rate
DB study
The primary efficacy end point was the proportion of patients treated with FKB327 versus the RP achieving ACR20 response rates at Week 24. The proportion of patients in the FKB327 group achieving an ACR20 response at Week 24 was within the predefined equivalence parameters recommended by the EU CHMP and the FDA.9 Therefore, equivalence was concluded between FKB327 and the RP.
Open-label extension
The group of patients rerandomised to FKB327 in Period 2 had a slightly lower ACR20 response rate at baseline/Week 0 (75.6%, n=245/324) compared with those rerandomised to the RP (81.9%, n=262/320); however, by Week 30, ACR20 response rates were similar for FKB327 (84.1%, n=233/277) and the RP (83.6%, n=240/287). In both groups, the overall ACR20 response rate decreased slightly over time from 78.7% (n=507/644) achieved at Week 0 (ie, Week 24 of the DB study) to 73.4% (n=473/644 at Week 30. ACR20 responses were evaluated to determine whether switching from the RP to FKB327 and vice versa had any effect on efficacy in Periods 2 and 3.
Over the 30-week period, the ACR20 response rate ranged from 77.5% (n=165/213) to 82.6% (n=176/213) in the RP group; for those who switched to FKB327, the response rate ranged from 73.1% (n=79/108) to 75.9% (n=82/108). Over the 30-week period, the ACR20 response rate ranged from 71.3% (n=154/216) to 75.5% (n=163/216) for patients who remained on FKB327; the response rate ranged from 67.6% (n=83/108) to 82.4% (n=89/108) for those who switched to the RP.
At the beginning of Period 3 (Week 30), ACR20 response rates were similar for all four treatment sequences. During Period 3, ACR20 response rates ranged from 75.7% (n=140/185) to 84.3% (n=156/185) for F-F-F, from 83.7% (n=77/92) to 88.0% (n=81/92) for RP-F-F and from 83.6% (n=158/189) to 86.8% (n=164/189) for RP-RP-F. Importantly, ACR20 response rates for patients in the F-RP-F sequence (double switch) were similar to other treatment groups, ranging from 74.5% (n=73/98) to 78.6% (n=77/98).
Secondary efficacy end points
The secondary efficacy end points included DAS28-CRP; ACR50/70 response rates; and TJC, SJC, CRP, visual analog scale scores, Health Assessment Questionnaire-Disability Index (HAQ-DI), erythrocyte sedimentation rate (ESR) and DAS28-ESR. The 95% CI of DAS28-CRP at Week 24 in the DB study was within the predefined limits of ±0.6, confirming equivalence.9 DAS28-CRP was similar in the FKB327 and RP groups at all visits from the start of the DB study to the OLE (figure 3). Mean DAS28-CRP at baseline of the DB study (>5.1) and OLE indicated high disease activity. Patients rerandomised to FKB327 in Period 2 had higher DAS28-CRP at baseline/Week 0 (3.53) than those rerandomised to the RP (3.40). Between weeks 0 and 30 of the OLE, mean DAS28-CRP decreased similarly for all patients, regardless of switching or maintaining treatment between studies. Mean DAS28-CRP ranged from 6.11 at baseline of the DB study to 3.13 at Week 30 for patients who maintained treatment with the RP (RP-RP, N=213), whereas the scores ranged from 5.99 at baseline of the DB study to 3.20 at Week 30 for those who switched to FKB327 (RP-F, N=108). For patients who maintained treatment with FKB327 throughout the DB study and Period 2 (F-F, N=216), mean DAS28-CRP ranged from 6.02 at baseline of the DB study to 3.04 at Week 30; for those who switched to the RP (F-RP, N=108), the scores ranged from 6.12 at baseline of the DB study to 3.28 at Week 30. For patients in the F-F and RP-RP sequences, similar decreases in mean DAS28-CRP were observed between baseline of the DB study and Week 30, and between weeks 0 and 80.

{kind=link}
{kind=link}
{kind=link}
ACR20 Response Rate and DAS28-CRP. ACR, American College of Rheumatology; DAS28-CRP, Disease Activity Score in 28 joints using C-reactive protein; F, FKB327; RP, reference product.
At the start of Period 3 (Week 30), DAS28-CRP was similar for all four treatment sequences. During Period 3, mean decreases in DAS28-CRP were similar in all four sequences until Week 76. Importantly, no difference in mean DAS28-CRP was observed for patients who switched from FKB327 to RP and back to FKB327 (F-RP-F; double switch) compared with the remaining three sequences.
As with the ACR20 response rate, patients rerandomised to FKB327 in the OLE had slightly lower baseline/Week 0 ACR50/70 response rates than those who received the RP. The ACR50 response rate at Week 0 of the OLE was 47.7% and 50.3%, respectively, for the FKB327 and RP groups, and increased at Week 30 to 58.5% and 59.2%, respectively. The ACR70 response rate for the FKB327 and RP groups was 21.0% and 24.7%, respectively, at Week 0, and 34.3% and 37.2%, respectively, at Week 30. The proportion of ACR50/70 responders increased similarly in both groups at all visits. Efficacy was maintained in all treatment groups and no effect of switching was evident from sequence comparisons.
The changes in all ACR core set variables were similar for both treatment groups. At Week 0 of the OLE, mean HAQ-DI values were lowest in the F-F sequence (1.19) and highest in the RP-F sequence (1.32). The reduction in HAQ-DI observed between baseline of the DB study and Week 0 was sustained from Week 0 to Week 30. The reduction in HAQ-DI was also observed and sustained from Week 30 to Week 54, but from Week 66, the data became more variable after longer-term follow-up. The mean DAS28-ESR decreased similarly in the FKB327 and RP groups, and was consistent with the decreases seen in DAS28-CRP.
Pharmacokinetics
DB study
The PK Analysis Set included 722 patients: 364 patients (99.2%) and 358 patients (98.6%) in the FKB327 and RP groups, respectively.9 The between-patient variability in systemic exposure to adalimumab was comparable between treatments.
Open-label extension
The mean serum drug concentrations at Week 0 were higher in the sequence groups that had been administered FKB327 in the DB study (F-F-F, 6500 ng/mL; F-RP-F, 6000 ng/mL; RP-F-F, 5170 ng/mL; RP-RP-F, 5720 ng/mL). The between-patient variability in systemic exposure was high throughout Period 2, with coefficient of variant ranging from 60.7% to 78.2% across the sequences. In all the sequences, the interindividual variability was high; however, the mean serum drug concentration appeared generally stable between Week 0 and Week 30 for the F-F-F (6000 ng/mL), RP-F-F (5730 ng/mL) and RP-RP-F (5750 ng/m) treatment sequences; a slight downward shift was observed for the F-RP-F (4790 ng/mL) sequence. Concentrations in patients who did not switch treatment compared with the F-F-F and RP-RP-F sequences remained stable and comparable during Period 2. Moreover, the time course of mean serum drug concentrations across Periods 2 and 3 by treatment sequence shows that serum drug concentrations were generally stable in all sequences.
Immunogenicity
DB study
At each sampling time point, the frequency of ADAs was comparable between treatments.9 The distribution of titre results at the last sampling point was comparable between treatments, with no differences in the proportions of patients in the FKB327 and RP groups at each positive ADA titre level. The proportion of patients with positive ADA activity was very similar in the FKB327 and RP groups (60.7% and 58.8% of patients, respectively) at the last sampling time point, with almost all samples testing positive for neutralising ADAs.
Open-label extension
The proportion of patients with positive ADA activity was highest at Week 0 and similar (approximately 60%) across treatment sequences (table 4). Mean ADA titre was higher for FKB327 and the RP at Week 30 compared with baseline, and was higher for the RP than for FKB327 at Week 30. Median ADA titre did not increase over time from Week 0 to Week 30 for both FKB327 and the RP. At Week 30 (end of Period 2), 51.9% of patients in the F-F-F sequence and 50.5% of patients in the RP-RP-F sequence with samples positive for ADA in the confirmatory assay tested positive for neutralising ADAs (titre ≥0.25). For the F-RP-F sequence, 60.0% of patients with samples positive for ADA in the confirmatory assay tested positive for neutralising ADAs. For the RP-F-F sequence, 45.2% of patients with samples positive for ADAs in the confirmatory assay tested positive for neutralising ADAs. At Week 80 (end of study), the percentage of patients with samples positive for ADA in the confirmatory assay testing positive for neutralising ADAs ranged from 41.8% in the RP-RP-F sequence to 55.2% in the F-RP-F sequence. Neutralising ADAs did not increase in any sequence during the study.
Summary of antidrug antibody status: safety analysis set
DISCUSSION
In the phase 3, multicentre, randomised, DB, parallel-arm, active-comparator, efficacy equivalence study, FKB327 or the RP was administered EOW for 22 weeks in patients with active RA whose disease was not controlled on MTX alone. The OLE was designed to compare the longer-term safety, efficacy, immunogenicity PK and multiple-dose PK of FKB327 with the RP for an additional 30 weeks of treatment (to a total of 1 year from the start of the preceding study), and to provide longer-term data for up to 76 weeks (total of 2 years) of treatment with FKB327. Results from the first 54 weeks (DB study and Period 2 of the OLE) have been previously published.9
The study was also designed to assess the effect on safety of switching from the RP to FKB327 and vice versa, by comparison of the four treatment sequences. The safety profile of FKB327 in this study is consistent with the known risks of the RP.3 4 The DB study demonstrated that the safety profiles of FKB327 and the RP were comparable in patients with moderate-to-severe, active RA for 24 weeks of exposure. In the OLE study, the overall exposure in patient-years was nearly four times greater for FKB327 (673.7) compared with the RP (175.4), due to patients switching from the RP to FKB327 for Period 3; therefore, the safety analysis focuses on IRs adjusted by overall exposure. The most commonly reported TEAEs were nasopharyngitis and other infections, which was previously reported with the RP and other biosimilar products.7 8 12 The incidence of ISRs, which are known side effects of adalimumab, was numerically lower with FKB327 compared with the RP. The difference in excipients between FKB327 and the RP, notably, the absence of citrate in FKB327, may have contributed to the observed decreased incidence of ISRs. A recent study investigating an etanercept biosimilar reported no ISRs in patients who received either the biosimilar or the RP, further supporting the safety of anti-TNF biosimilars.13 The results suggest no difference in the safety profile between FKB327 and the RP, and no change in AE profile for longer-term FKB327 treatment.
The results showed no evidence to indicate that switching between treatments has any impact on safety. No effect of switching treatments was seen in Period 2, and data from Period 3 did not indicate any effect of double-switching treatment. This study is the first to report double switching of treatment between an adalimumab biosimilar and the originator, which suggests no increased risk for multiple switching in patients with RA, similar to observations with an etanercept biosimilar treatment for psoriasis.14 In the EGALITY study, patients underwent three treatment switches at 6-week intervals, after which patients were followed through Week 52, which was shorter than the 6-month switching interval and 2-year follow-up in the current study.
The impact of ADA titre on the serum trough concentration for FKB327 and the RP was comparable between treatment groups. The proportion of patients with positive ADA activity was highest at Week 0, was similar (approximately 60%) across the sequences and was higher than reported in other studies with adalimumab biosimilars (16%–18%8, 38%12–50%7). This may be due to differences in the sensitivity of the assay used in each study. The proportion of patients with positive ADA status decreased between Week 0 and Week 30, and switching treatment from the RP to FKB327 or vice versa was not associated with a discernible influence on ADA response. At Week 30, the majority of patients with samples positive for ADAs in the confirmatory assay tested positive for neutralising ADAs (titre ≥0.25); therefore, almost all the ADAs identified by the confirmatory assay were shown to be neutralising.
The efficacy profile was comparable for FKB327 and the RP over the first 30 weeks of the study. Data from patients who switched treatment after rerandomisation to the OLE, particularly from the RP to FKB327, provide robust information regarding the use of biosimilar products in clinical practice. Data from patients who switched treatment twice may also support future interchangeability applications in the United States. Switching from the RP to FKB327 was not associated with loss of efficacy or increases in TEAEs or immunogenicity. No effect of switching treatments was seen in Period 2 of this study, and data from Period 3 did not indicate any effect of double-switching treatment. No meaningful differences were observed in safety, immunogenicity or efficacy for patients who switched from the RP in the DB study to FKB327 in the OLE or vice versa. Similar results of treatment safety, efficacy and immunogenicity were reported in the phase 3 EGALITY study of an etanercept biosimilar in patients with chronic plaque psoriasis14; a phase 3 study of a rituximab biosimilar in patients with RA15; and a phase 3 study of an infliximab biosimilar in patients with active Crohn’s disease.16 Both the EGALITY study and the current study assessed multiple switching between biosimilars and RP treatment; however, the EGALITY study had shorter treatment intervals and follow-up.14 Studies investigating the rituximab and infliximab biosimilars included a single-switch treatment with follow-up to 72 or 54 weeks, respectively, which was shorter than in the current study.15 16 Although these studies reported similar outcomes, the current data, based on 6-month switching intervals and 2-year follow-up, provide more robust clinical information on the safety and efficacy of treatment switching in patients with RA.
A secondary objective of the OLE was to compare the efficacy of maintenance treatment with FKB327 and the RP after induction of a response in the DB study. The key variables were ACR20/50/70 response rates and DAS28-CRP. Some imbalances in baseline disease characteristics were observed once patients were rerandomised, and baseline disease activity was higher in patients allocated to FKB327 compared with those receiving the RP. However, ACR20/50/70 response rates increased slightly on both treatments up to Week 30, with no difference observed at the end of Period 2. Similar decreases in mean DAS28-CRP and individual ACR core set variables were observed over Period 3 with FKB327 and the RP.
The study had limitations to consider, including the open-label nature of the extension period. All patients were on active drug, which may have increased perceived responses and expectation of improvement. Because patients were required to complete the Week 24 visit of the randomised DB study to enroll in the OLE, the potential existed for selection bias. Survival bias may have existed in the OLE, and patients experiencing AEs or poor response may have discontinued, whereas those who responded well continued in the study, possibly inflating the efficacy responses. To combat this artificial increase in response rates, the European League Against Rheumatism recommends the use of absolute numbers for data analysis.17
Previous studies have reported subjective health complaints that may be due to the nocebo response, or patients’ own negative expectations that lead to negative symptoms during treatment.18 19 In the current study, no clear difference in AEs was reported among patients in different treatment sequences, and the discontinuation rate did not reflect a clear pattern based on receipt of FKB327 or the RP.
This report provides evidence that double-switching treatment had no effect on safety; however, the number of evaluable double-switched patients was limited, and the study design allowed for evaluation of double switching for patients in the F-RP-F treatment group, but not in those in the RP-F-RP group; however, F-RP-F is to be expected in clinical practice. Further investigation is needed for switching treatments between the reference and biosimilar products.11
Importantly, the results of the current study significantly extend the preliminary analysis conducted after the first 54 weeks.9 The preliminary analysis demonstrated comparability of FKB327 and RP in terms of efficacy, safety and immunogenicity. Furthermore, no effect of switching was observed between treatments in the OLE in the preliminary analysis through Week 54. The current report, which is the final analysis of the 2-year DB study and OLE, provides long-term results, extending the previously published results out to 2 years and demonstrating comparable safety, efficacy and immunogenicity of FKB327 with the RP. Furthermore, no effect of single or double switching between treatments was reported in this longer-term setting.
CONCLUSION
Data from the DB and OLE studies demonstrate that the safety and efficacy of the adalimumab biosimilar FKB327 were maintained and comparable to RP adalimumab, both over long-term treatment and when patients were switched between the biosimilar and the RP. Similarly, immunogenicity was comparable between FKB327 and the RP over long-term treatment and in the switching portions of the study. Because these results support the biosimilarity of FKB327 and the RP over a period of 2 years, with no impact of switching and double switching between the two drugs, FKB327 should be useful in all chronic immune-mediated inflammatory diseases for which adalimumab is indicated.
Acknowledgments
Funding for the double-blind study, FKB327-002 (NCT02260791), and open-label extension study, FKB327-003 (NCT02405780), was sponsored by Fujifilm Kyowa Kirin Biologics Co. Technical editorial and medical writing assistance was provided under the direction of the authors by Strategix, an affiliate of The Lynx Group LLC; funding for this support was provided by Mylan Inc. Statistical expertise and the primary statistical analysis were provided by Takahiro Ito, senior manager, Biostatistics, at Kyowa Kirin Pharmaceutical Development.
Footnotes
Contributors MG, RA and YA initiated the study design and helped with implementation. MG and RA are grant holders. MG, RA, YA and RM contributed to the refinement and development of the manuscript and approved the final manuscript.
Competing interests MG has received consultant fees from Fujifilm Kyowa Kirin Biologics Co., including for the design of this trial. HK has no conflicts of interest to report. YA is an employee of Fujifilm Kyowa Kirin Biologics Co. and has received personal fees from the company, both during the conduct of the study and outside the submitted work. RM is an employee and shareholder of Mylan Inc. RA has received consultant fees from Mylan Inc and has been a paid speaker for Mylan Inc.
Patient consent for publication Consent obtained directly from patient(s).
Disclaimer The authors were fully responsible for all content and editorial decisions and received no financial support or other form of compensation related to the development of this manuscript.
Data sharing statement Data are available in a public, open-access repository.
Provenance and peer review Not commissioned; externally peer reviewed.