Article Text

Abstract
Objective Standard assessment of interferon (IFN) system activity in systemic rheumatic diseases depends on the availability of RNA samples. In this study, we describe and evaluate alternative methods using plasma, serum and DNA samples, exemplified in the IFN-driven disease primary Sjögren’s syndrome (pSS).
Methods Patients with pSS seropositive or negative for anti-SSA/SSB and controls were included. Protein-based IFN (pIFN) scores were calculated from levels of PD-1, CXCL9 and CXCL10. DNA methylation-based (DNAm) IFN scores were calculated from DNAm levels at RSAD2, IFIT1 and IFI44L . Scores were compared with mRNA-based IFN scores measured by quantitative PCR (qPCR), Nanostring or RNA sequencing (RNAseq).
Results mRNA-based IFN scores displayed strong correlations between B cells and monocytes (r=0.93 and 0.95, p<0.0001) and between qPCR and Nanostring measurements (r=0.92 and 0.92, p<0.0001). The pIFN score in plasma and serum was higher in patients compared with controls (p<0.0001) and correlated well with mRNA-based IFN scores (r=0.62–0.79, p<0.0001), as well as with each other (r=0.94, p<0.0001). Concordance of classification as ‘high’ or ‘low’ IFN signature between the pIFN score and mRNA-based IFN scores ranged from 79.5% to 88.6%, and the pIFN score was effective at classifying patients and controls (area under the curve, AUC=0.89–0.93, p<0.0001). The DNAm IFN score showed strong correlation to the RNAseq IFN score (r=0.84, p<0.0001) and performed well in classifying patients and controls (AUC=0.96, p<0.0001).
Conclusions We describe novel methods of assessing IFN system activity in plasma, serum or DNA samples, which may prove particularly valuable in studies where RNA samples are not available.
- Sjögren’s syndrome
- interferon
- IFN
- Nanostring
- DNA methylation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Excessive interferon system activation is a common feature in several systemic autoimmune diseases and standard assessment of interferon system activity depends on the availability of RNA samples.
What does this study add?
We present two novel methods enabling assessment of interferon system activity in plasma, serum and DNA samples.
How might this impact on clinical practice?
The presented methods enable assessment of interferon system activity in research studies and historical cohorts where RNA samples are not available.
Introduction
Systemic autoimmune diseases are characterised by abnormal immunological targeting of self-tissue, resulting in dysregulated inflammation and multiorgan damage. Uncontrolled immune activation can be observed with immune complex deposition and leucocyte infiltration of affected tissues, as well as skewed immune cell populations and high levels of proinflammatory cytokines in the peripheral circulation. Specifically, excessive activation of the interferon (IFN) system is a well-established phenomenon in several systemic autoimmune diseases, such as systemic lupus erythematosus (SLE) and primary Sjögren’s syndrome (pSS).1 2
The numerous subtypes of type I IFNs and their low concentrations in plasma make it difficult to develop assays to measure them. Although a recent report showed encouraging results of direct measurement of IFN-α in plasma,3 a commercially available assay for robust quantification of type I IFNs is lacking. Therefore, activity of the IFN system is commonly assessed by measuring expression of IFN-stimulated genes, whereby mRNA-based IFN scores are calculated.
The contribution of IFNs to the pathology of systemic autoimmune diseases is well documented.1 2 Indeed, the importance of IFN-α is evidenced by the development of SLE or SLE-like symptoms in patients receiving treatment with IFN-α.4 5 In pSS, a majority of patients present with high mRNA expression-based IFN scores, which have been associated with increased disease activity, high frequency of Ro/SSA and/or La/SSB autoantibodies, high IgG levels and high gene expression of B cell activating factor in monocytes.6
The IFN system has long been considered a promising therapeutic target, and monoclonal antibodies targeting either IFN-α or the IFN alpha/beta receptor 1 (IFNAR1) have been developed.7 Anifrolumab, a monoclonal antibody targeting IFNAR1 substantially reduced disease activity in patients with SLE in a phase II clinical trial, with greater effect size in patients with high IFN signature at baseline.8 Thus, careful subclassification of patients who present with high IFN scores and therefore may benefit more from IFN blockade could be essential in future clinical trials. Moreover, the IFN score has the potential to function as a marker in monitoring treatment responses and for aiding physicians in clinical decisions.
In this study, we present alternative methods of assessing IFN system activity. We show that quantification of IFN activity can be achieved through measurement of IFN-induced proteins compiled into a protein IFN (pIFN) score in plasma or serum. Furthermore, we describe a novel method of assessing IFN system activity in DNA methylation (DNAm) data through calculation of a DNAm IFN score, thus enabling insight between genotype and phenotype. Importantly, these methods may prove particularly valuable in studies where RNA samples are not available, which is often the case in large cohort studies.
Methods
Study population
The study included a total of 302 patients fulfilling the American-European consensus group criteria for pSS9 and 684 healthy controls (HC). All patients were diagnosed and sampled and all controls recruited and sampled at the Karolinska University Hospital or Uppsala University Hospital. Monocytes and B cells were available from 25 patients and 16 HC, for which also plasma (all) and serum (the majority) were available (table 1). B cells for DNAm analysis were available from 24 patients and 47 HC, out of which RNA-sequencing (RNAseq) data were available for coanalysis for 16 patients and 20 HC (table 1). Whole blood was available for DNAm analysis from 75 anti-SSA positive, 25 anti-SSA/SSB negative patients with pSS and 587 HC. Serum alone was available for 83 anti-SSA positive and 87 anti-SSA/SSB negative patients with pSS, and 34 HC. No included patient was positive for anti-SSB alone, wherefore patients with anti-SSA with/without anti-SSB as a group were denoted anti-SSA positive.
Clinical characteristics of patients with pSS and healthy controls from whom samples were used for correlation analyses
Isolation of cells from peripheral blood and plasma collection
For quantitative PCR (qPCR) and Nanostring, CD19+ B cells and CD14+ monocytes were isolated from peripheral blood mononuclear cells (PBMCs) obtained using Vacutainer CPT Tubes (BD Biosciences), followed by magnetic microbead sorting in an autoMACS Pro Separator (Miltenyi Biotec). The autoMACS cell purity was sampled by flow cytometry (Gallios, Beckman Coulter) and median purity was found to be 98.5% for B cells and 95.3% for monocytes. For DNAm analysis and RNAseq, B cells were isolated from PBMCs obtained applying Ficoll-Hypaque density-gradient centrifugation (GE Healthcare) followed by magnetic microbead sorting (Miltenyi Biotec). Purity >95% was confirmed by sampling via flow cytometry (FACSCanto II, BD Biosciences). Plasma and serum samples were frozen at −80°C within 2 hours of venipuncture.
Isolation of RNA and gene expression analyses
For qPCR and Nanostring, RNA was extracted using RNeasy Mini Kits (Qiagen). For RNAseq and DNA preparation, the AllPrep DNA/RNA Mini Kit and the AllPrep DNA/RNA/miRNA Universal Kit (Qiagen) were used.
Expression of 594 genes was measured (Immunology CodeSet v2, NanoString Technologies). Data were quality controlled and normalised using the nSolver Analysis Software (V.3.0.22, NanoString Technologies). Expressions of MX1, STAT1, IRF7, IFITM1 and IFI35 were used to calculate Nanostring IFN scores.
Reverse transcription was performed using the iScript cDNA Synthesis Kit (Bio-Rad). PCR was performed with a CFX384 real-time system using iQ SYBR Green Supermix (Bio-Rad) and a 2-step protocol (95°C 3 min, 95°C 10 s and 60°C 45 s for 40 cycles). Primer sequences are presented in online supplementary table 1. Gene expression was related to β-Actin calculated by the CFX Manager software (Bio-Rad). Expressions of IFI44L, IFI44, IFIT3, LY6E and MX1 were used to calculate qPCR IFN scores.6
Supplemental material
RNAseq
RNAseq was performed as previously described.10 In summary, the TruSeq stranded mRNA sample preparation kit was used for library preparation followed by paired-end sequencing with 50 bp read length on a (Illumina). Data were quality controlled using the RNA-SeQC pipeline11 and alignment of sequencing reads to the human reference genome (build GRCh37) was performed with TopHat2 (2.0.4).12 Expression levels were normalised to fragments per kilobase of exon per million fragments mapped within the Cufflinks framework.13 Expressions of MX1, STAT1, IRF7, IFITM1 and IFI35 were used to calculate RNAseq IFN scores.
Proximity extension assay
Levels of 92 plasma and serum proteins were measured by proximity extension assay (PEA) technology (Immuno-Oncology panel, Olink Bioscience).14 Samples were analysed and data were quality controlled and normalised at the core Clinical Biomarkers Facility at Science for Life Laboratory, Uppsala, Sweden.
DNAm analysis
DNAm was analysed as described previously.15 Briefly, DNAm of 485 577 CpG sites was determined using the HM450K BeadChip (Illumina). Signal intensities were parsed into the Minfi R package16 for quality control and Subset-quantile Within Array Normalisation.17 Methylation beta values were calculated as the fraction of the signal intensity from the methylated CpG sites over the total intensity (range 0–1, corresponding to 0%–100% methylation). DNAm beta values at type I IFN-regulated genes with the strongest correlation with the RNAseq IFN score were identified at cg10549986 (RSAD2), cg05552874 (IFIT1) and cg05696877 (IFI44L) and were used to calculate DNAm IFN scores.
Calculation of scores
IFN-regulated genes were identified through the Interferome V.2.01 database.18 Scores were calculated according to a previously described formula.19 The mean (meanHC) and SDHC for each gene, protein or methylation level in the HC group were used to achieve standardised values (SVs) for each individual according to the formula: SV=(Value−MeanHC)/SDHC. Subsequently, SVs were summed up to total scores.
Statistical analysis
Mann-Whitney U test was used to compare two groups and Spearman’s rank correlation coefficient was used when assessing statistical association between two variables. Kruskal-Wallis with post hoc Dunn’s test was used when comparing more than two groups. Receiver operating characteristic (ROC) curves were generated to establish thresholds for IFN score positivity and to obtain area under the curve (AUC) values. Statistical tests were performed using GraphPad Prism (V.6.0, GraphPad). P values <0.05 were considered significant.
Results
Correlation between qPCR and Nanostring mRNA-based IFN scores
To establish mRNA-based IFN scores, qPCR of five type I IFN-regulated genes was performed on RNA from B cells and monocytes from patients with pSS (n=25) and HC (n=16) as previously described.6 One monocyte qPCR sample was excluded due to poor sample quality. As expected, the qPCR IFN scores were significantly higher in patients compared with HC for both B cells and monocytes (figure 1A,D). Likewise, mRNA-based IFN scores were calculated from the expression of five type I IFN-regulated genes measured by Nanostring, and also revealed higher mRNA IFN scores in patients compared with HC (figure 1B,E). Of note, one HC consistently displayed high IFN scores throughout the analyses (figure 1A–F).

mRNA-based interferon (IFN) scores analysed by qPCR and Nanostring reveal high IFN scores in patients with primary Sjögren’s syndrome (pSS) compared with healthy controls (HCs) and correlation between the methods. (A, B) IFN scores were calculated from gene expression levels of five IFN-regulated genes in B cells from patients with pSS and HC. (C) Correlation analysis of IFN scores analysed by qPCR and Nanostring in B cells. (D, E) IFN scores were calculated from gene expression levels of five IFN-regulated genes measured in monocytes from patients with pSS and HC. (F) Correlation analysis of IFN scores analysed by qPCR and Nanostring in monocytes. (G, H) Correlation analysis of IFN scores measured by qPCR and Nanostring in simultaneously sampled B cells and monocytes. (A, B, D, E) Dotted lines indicate thresholds for IFN signature positivity as identified by receiver operating characteristic curve analysis. (C, F, G, H) Spearman’s rank correlation coefficient. Horizontal bars represent mean±SEM. ****P<0.0001, Mann-Whitney U test. qPCR, quantitative PCR; SEM, SE of the mean.
To compare performance of the qPCR and Nanostring methods, correlation analysis was performed on the IFN scores in B cells and in monocytes, respectively. The analysis revealed a strong correlation between the two scores both in B cells (r=0.92, p<0.0001) and in monocytes (r=0.92, p<0.0001) (figure 1C,F). In all, these data demonstrate that both qPCR and Nanostring technology can be applied to establish mRNA-based IFN scores in peripheral B cells and monocytes with comparable performance.
mRNA IFN scores in peripheral blood B cells and monocytes are comparable
To better understand how well IFN scores in peripheral B cells correspond to IFN scores in monocytes, correlation analysis was performed on the scores determined by qPCR and by Nanostring. The analysis revealed a high degree of correlation between the IFN scores, both when analysed by qPCR (r=0.93, p<0.0001) and by Nanostring (r=0.95, p<0.0001) (figure 1G,H). These data reveal similar activation of IFN-regulated genes in simultaneously sampled peripheral B cells and monocytes demonstrating that either of these cell types may be used for determining IFN scores with comparable results.
A plasma pIFN score can be used to assess IFN system activity
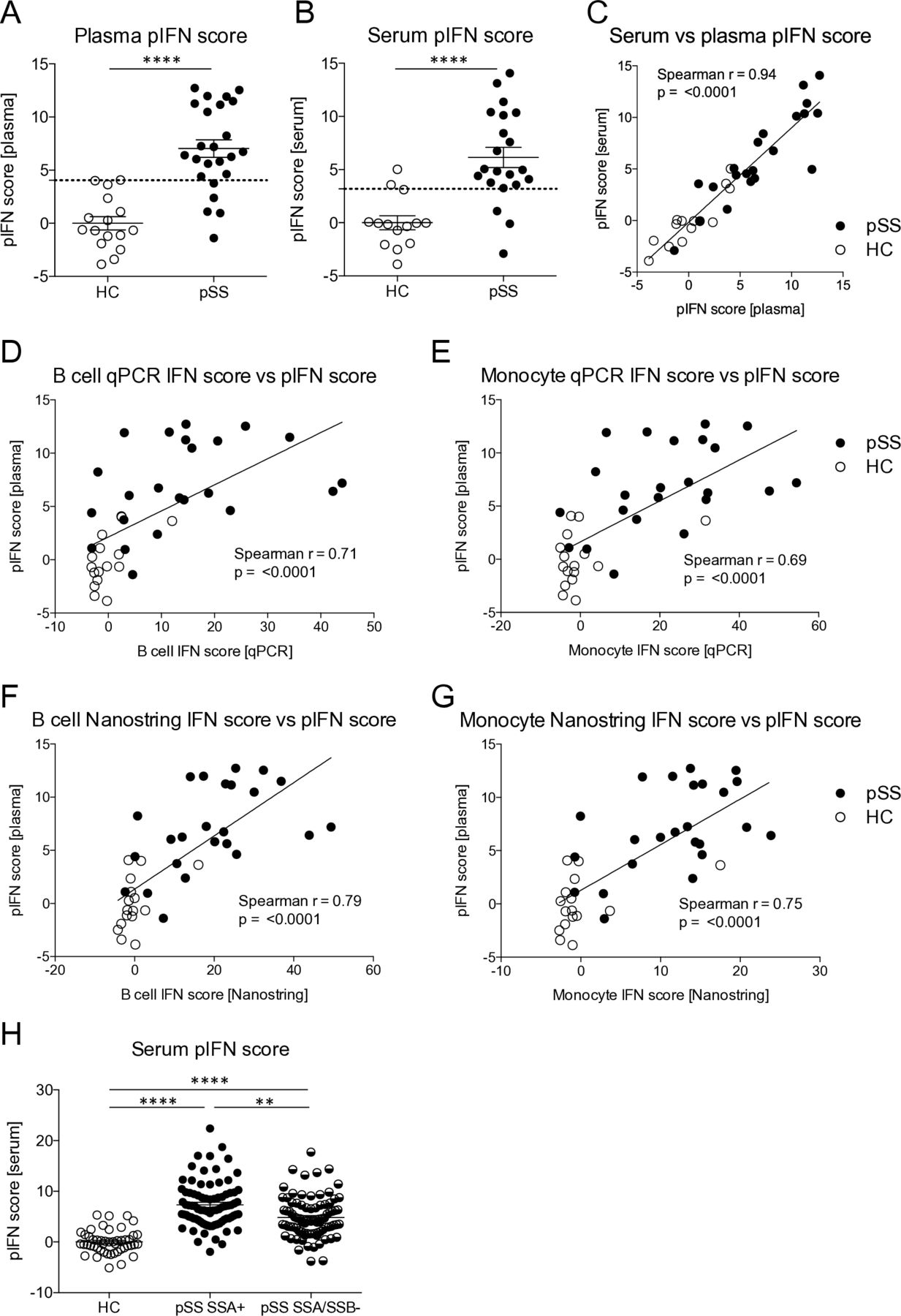
Plasma and serum samples are often more readily available for existing cohorts than RNA samples, and may, therefore, be useful for assessment of an IFN signature. We hypothesised that a plasma or serum pIFN score would reflect IFN system activity in a similar way as the mRNA-based IFN score. To determine which proteins that may be used for calculation of such a score we employed PEA, a novel technology using high-sensitivity dual antibody recognition of 92 proteins.14 One plasma sample had to be excluded due to failed analysis. Levels of plasma proteins identified as type I IFN regulated at the mRNA level in the Interferome V.2.01 database18 were correlated to qPCR IFN scores in B cells and the top three proteins with the strongest correlation (the soluble form of programmed cell death protein 1 sPD-1, CXCL10 and CXCL9) were used for calculation of a pIFN score (online supplementary table 2, online supplementary figure 1A–C). Patients displayed higher pIFN scores compared with HC in both plasma and serum (p<0.0001), with a high degree of correlation between the two (r=0.94, p<0.0001) (figure 2A–C). Furthermore, pIFN scores correlated well with mRNA-based IFN scores calculated in B cells and in monocytes by qPCR and Nanostring. Data on these correlations are visualised for plasma in figure 2D–G displaying the strongest association with the B cell Nanostring IFN score (r=0.79, p<0.001); the correlation coefficients for the serum pIFN score ranged between r=0.62–0.74 with p<0.0001 for all, data not shown. The serum pIFN score calculated in an extended set of patients (n=170) confirmed significantly higher pIFN scores in anti-SSA positive patients compared with anti-SSA/SSB negative patients (figure 2H). These data suggest that a pIFN score calculated from plasma or serum levels of sPD-1, CXCL9 and CXCL10 is a valid alternative to mRNA-based IFN scores.
Supplemental material
Supplemental material

The protein interferon (pIFN) score measured in plasma or serum is a surrogate marker for IFN system activity. (A, B) pIFN scores were calculated from protein levels of PD-1, CXCL9 and CXLC10 measured by proximity extension assay technology in plasma and serum from patients with primary Sjögren’s syndrome (pSS) and healthy controls (HCs). Dotted lines indicate thresholds for IFN signature positivity as identified by receiver operating characteristic curve analysis. (C) Spearman’s rank correlation coefficient between the pIFN scores measured in simultaneously sampled serum and plasma samples from the same individuals (n=35). (D–G) Spearman’s rank correlation coefficient between the pIFN score and mRNA-based IFN scores measured (D, E) by qPCR and (F, G) by Nanostring in B cells and monocytes. (H) Serum pIFN score measured by proximity extension assay in HC (n=48), and in patients with pSS positive for anti-SSA (SSA+, n=83) or negative for anti-SSA/SSB (SSA/SSB-, n=87) autoantibodies. Horizontal bars represent mean±SEM. (A, B) ****P<0.0001, Mann-Whitney U test. (H) **P<0.01, ****P<0.0001, Kruskal-Wallis with post hoc Dunn’s test. qPCR, quantitative PCR; SSA, Sjögren’s syndrome antigen A; SSB, Sjögren’s syndrome antigen B; SEM, SE of the mean.
Performance and concordance of mRNA IFN scores and the pIFN score
When using IFN scores for patient stratification, a threshold is commonly used to define study subjects as IFN signature positive or negative.6 20 To assess concordance rates of the scores we had generated, ROC curves were used to determine thresholds for IFN signature positivity. Thresholds were selected to achieve the highest possible sensitivity and specificity. ROC curve analysis revealed the highest value for the AUC in B cells and monocytes analysed by Nanostring, as well as the plasma pIFN score (all AUC=0.93, p<0.0001), closely followed by the other IFN scores (AUC=0.88–0.89, p<0.0001) (figure 3A–F). Concordance rates for IFN score positivity were consistently high among the mRNA-based IFN scores (ranging from 95.0% to 100%), as well as for the pIFN scores (ranging from 79.5% to 88.6%) (table 2). In all, these data demonstrate that mRNA-based IFN scores as well as the pIFN score can accurately classify patients and controls. Additionally, high concordance rates between mRNA IFN and pIFN scores further support their potential use.
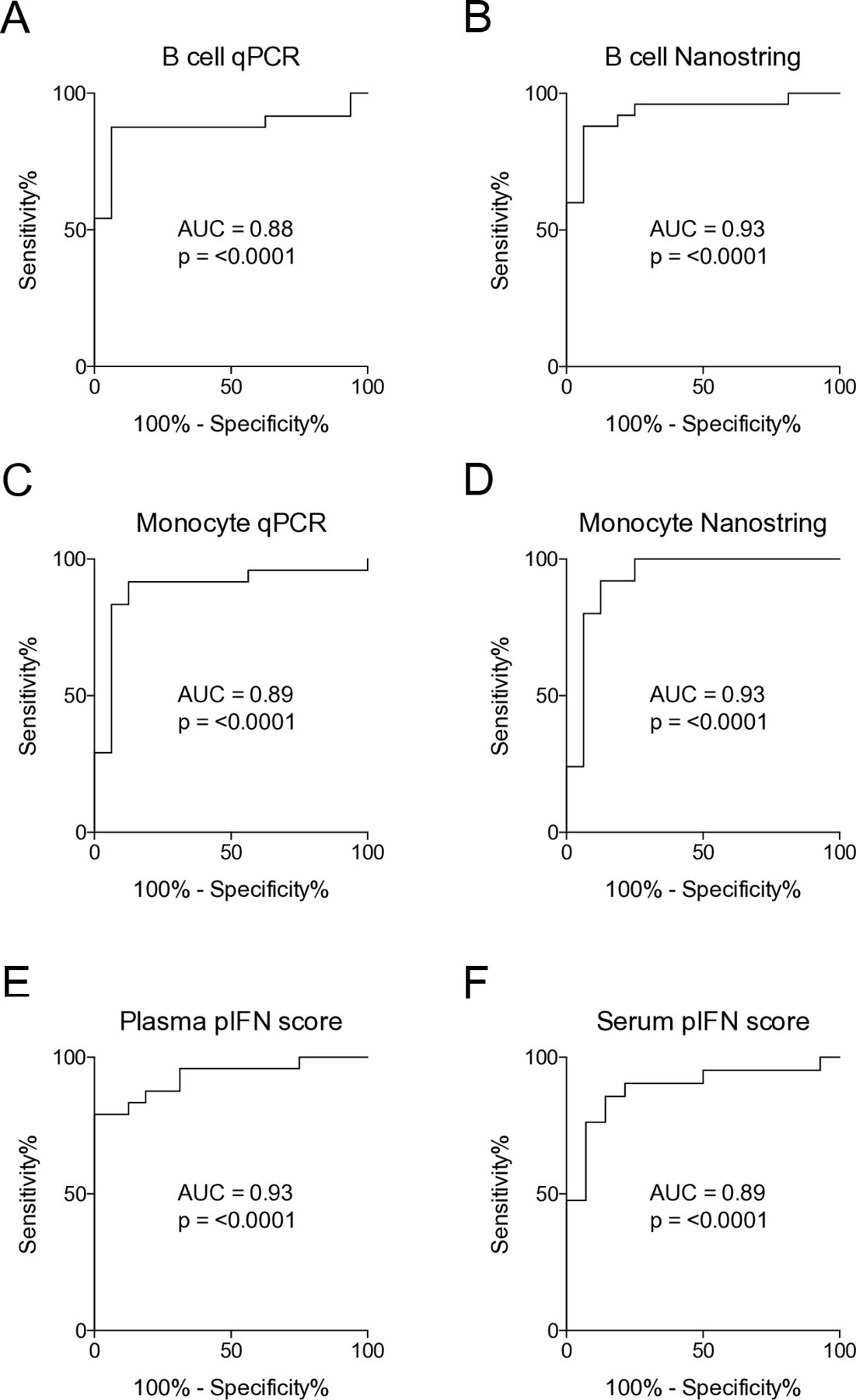
Receiver operating characteristic (ROC) curve analysis establishes thresholds for interferon (IFN) signature positivity. (A–F) ROC curve analysis of mRNA-based IFN scores measured by qPCR and by Nanostring, and the plasma and serum protein IFN (pIFN) scores. AUC, area under the curve; qPCR, quantitative PCR.
Concordance of IFN scores
Assessing level of IFN system activity by measuring DNAm
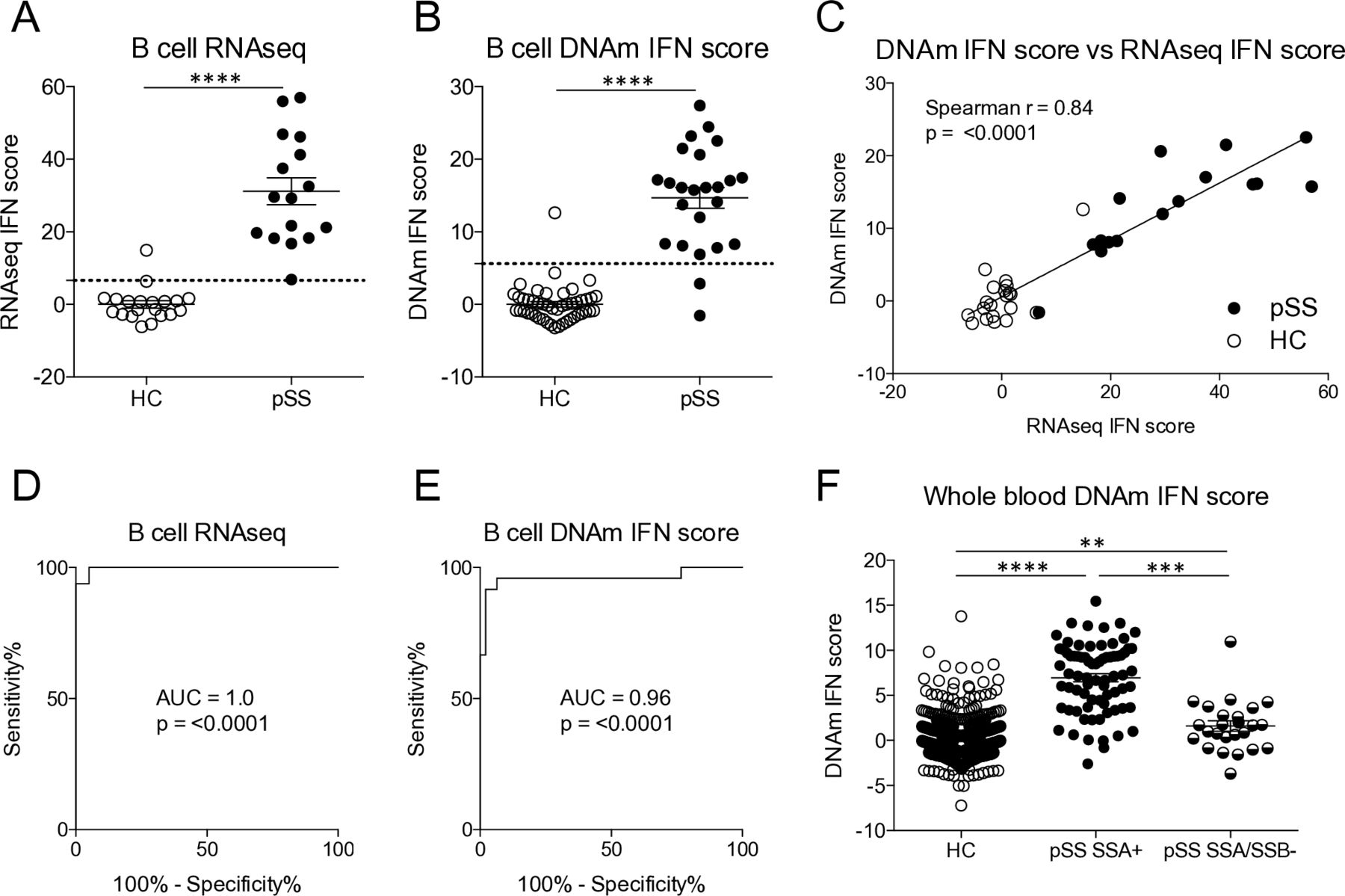
Having observed that a pIFN score correlates well with mRNA-based scores, we hypothesised that DNA may also be used for defining an IFN score. In many studies, DNA is the only biological material available for investigation, and a connection between genotype and phenotype may be of high value. To address this possibility, we first calculated an IFN score from RNAseq data. Thereafter, a DNAm IFN score was calculated from the beta values of the three CpG sites at type I IFN-regulated genes with the strongest correlation with the RNAseq IFN score. The RNAseq IFN and the DNAm IFN scores were higher in patients with pSS compared with HC (figure 4A,B), and both scores performed well at classifying patients and controls with an AUC=1.0, p<0.0001 for the RNAseq IFN score and an AUC=0.96, p<0.0001 for the DNAm IFN score (figure 4D,E). Moreover, the correlation between the two scores was high (r=0.84, p<0.0001) (figure 4C). Finally, using the threshold set by ROC curve analysis the concordance of IFN positivity was found to be 97.2% (table 2). The two scores only classified 1 out of 36 individuals differently. In a separate, larger cohort of patients (n=100) and controls (n=587), a DNAm IFN score calculated in whole blood was significantly higher in anti-SSA positive patients than in anti-SSA/SSB negative patients (figure 4F). In all, these data suggest that assessment of IFN activity may be performed on epigenetic CpG methylation data, with a high concordance with RNAseq IFN scores.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A DNA methylation interferon (DNAm IFN) score is higher in patients compared with controls and constitutes a surrogate marker for IFN system activity. (A) RNAseq IFN scores were determined based on the expression of five IFN-regulated genes in B cells from patients with primary Sjögren’s syndrome (pSS) and healthy controls (HCs). (B) DNAm IFN scores were calculated from methylation levels of three CpG sites in B cells located at the IFN-regulated genes RSAD2, IFIT1 and IFI44L. (C) Spearman’s rank correlation coefficient between the RNAseq IFN score and the DNAm IFN score measured in B cells. (D, E) Receiver operating characteristic (ROC) curve analysis was performed to establish thresholds for IFN signature positivity. (F) DNAm IFN scores were calculated in whole blood from HC (n=587), and in anti-SSA positive (SSA+, n=75) and anti-SSA/SSB negative (SSA/SSB-, n=25) patients with pSS. Dotted lines indicate thresholds for IFN signature positivity as identified by ROC curve analysis. Horizontal bars represent mean±SEM. (A, B) ****P<0.0001, Mann-Whitney U test. (F) **P<0.01, ***P<0.001, ****P<0.0001, Kruskal-Wallis with post hoc Dunn’s test. AUC, area under the curve; DNAm, DNA methylation; RNAseq, RNA sequencing; SSA, Sjögren’s syndrome antigen A; SSB, Sjögren’s syndrome antigen B; SEM, SE of the mean.
Discussion
Aberrant IFN system activation and its involvement in the immunopathology of systemic rheumatic diseases is well established.1 2 Currently, standard methods of assessing IFN system activity rely on gene expression analysis, which depends on the availability of RNA samples and that these have been well preserved. Thus, alternative methods for assessing IFN system activity would be of great value. In this study, we developed and evaluated two methods of measuring IFN system activity in plasma, serum and DNA samples. Additionally, we assessed the correlation of mRNA-based IFN scores measured by qPCR and Nanostring in simultaneously sampled cells of different lineages.
First, we measured mRNA-based IFN scores by both qPCR and Nanostring technology. We observed a strong correlation between IFN scores measured by the two methods, thus confirming previous reports that Nanostring is a valid alternative to qPCR for easy quantification of mRNA-based IFN scores.21 22
Second, we examined the correlation of mRNA-based IFN scores between monocytes and B cells. We observed strong correlations between IFN scores in the two cell types. In pSS, IFN signatures have previously been described in numerous cell types and tissues such as salivary glands,23 whole blood,24 PBMCs,25 B cells10 and monocytes.6 To our knowledge, our study is the first to examine the association of IFN scores between two different cell types in the blood of patients with pSS, showing that prototypic IFN-regulated genes initially described for calculation of IFN scores in either monocytes6 or B cells10 may be used for both cell types. These data are also in accordance with previous observations made in patients with SLE.26 27
Wanting to find a surrogate measure for IFN system activity in plasma and serum, we established a pIFN score based on the levels of three IFN-regulated proteins. Proteins were measured by PEA technology, which uses oligonucleotide-labelled antibody pairs for detection of analytes with high specificity and sensitivity.14 The pIFN scores displayed a good correlation with the mRNA-based IFN scores and performed well at classifying individuals as having ‘high’ or ‘low’ IFN signature with concordance rates to mRNA-based IFN scores ranging between 79.5% and 88.6%. Notably, the pIFN scores also displayed high accuracy at classifying patients and controls. The pIFN score was calculated from the levels of sPD-1, CXCL9 and CXCL10 (also denoted IP-10). PD-1 is a cell surface receptor involved in suppressing inflammatory activity. Notably, the PD-1/PD-L pathway has been implicated in autoimmunity,28 and there are reports of patients developing Sjögren’s syndrome during the treatment with drugs targeting the PD-1/PD-L pathway.29 A recent study from our group found increased expression of PD-1 on follicular regulatory T cells in patients with pSS.30 PD-1 also exists in a soluble form denoted sPD-1 which is thought to functionally antagonise its membrane bound form by blocking the PD-1/PD-L pathway.31 Interestingly, high levels of sPD-1 have been observed in the circulation of patients with systemic sclerosis and rheumatoid arthritis, also correlating with disease parameters.32–34 Thus, sPD-1 constitutes an interesting area for additional research. CXCL9 and CXCL10 are chemokines of the C-X-C chemokine family, which have both been reported to be upregulated at the mRNA and protein level in various tissues of patients with pSS.35–38 In fact, CXCL10 has previously been proposed as a surrogate marker for IFN activity in patients with SLE.39 40 In our study, compiling three proteins into one pIFN score resulted in optimal correlation and concordance with mRNA-based IFN scores. Reduced variability obtained from combining several proteins into one score should be beneficial in clinical applications. Addition of more than three proteins to the score did not improve its performance. Several plasma proteins or cell surface markers have been suggested as surrogate markers for IFN activity in systemic autoimmune diseases or interferonopathies such as galectin-9,41 SIGLEC1,42 MxA,43 CXCL1040 and CD64.44 However, analogous to mRNA-based IFN scores, we suggest that a combination of several IFN-regulated proteins compiled into one score may be advantageous to obtain robust results.
We also found that IFN activity could be assessed in DNAm data, and that such an approach generated scores with a very strong correlation with RNAseq IFN scores. Strikingly, out of 36 individuals, the DNAm IFN score only classified one individual differently into ‘high’ or ‘low’ IFN signature compared with the RNAseq IFN score. Previously, IFN-regulated genes have been reported to be hypomethylated in various tissues and cell types both in pSS15 and in SLE.45 46 The temporal relationship between hypomethylation of IFN-regulated genes and disease progression remains to be established. However, epigenetic modifications remain relatively stable over time which may suggest that the DNAm IFN score and pIFN score could be used for different purposes. We speculate that the DNAm IFN score may be better at correctly classifying patients and controls, while the pIFN may be more dynamic and thus more suitable for monitoring treatment responses.
We also investigated the performance of the pIFN and DNAm IFN scores in larger cohorts of patients. Both the serum pIFN score and DNAm IFN score calculated in whole blood were significantly higher in anti-SSA positive compared with anti-SSA/SSB negative patients. These data further corroborate that the suggested scores adequately measure IFN system activity, as IFN scores are known to be lower in seronegative patients.6 24
Limitations of the study include the relatively small number of individuals included with RNA samples available and that the IFN scores were only investigated in pSS. Also, the proteins constituting the pIFN scores were quantified by PEA which is a novel technology currently not widely spread. Quantification of the proteins by conventional methods such as ELISA could be preferable in possible future clinical routine applications, and we hope to address this in future studies.
In conclusion, we describe two novel methods of assessing IFN system activity in patients with pSS. We envision that these alternative protein and DNAm-based scores may prove useful as plasma, serum or DNA samples are often more readily available in large research studies and historically collected cohorts. Phenotypic subclassification of patients according to the degree of IFN system activity could prove to be of central importance as novel therapies are being developed and evaluated in clinical trials.
Acknowledgments
We thank Amina Ossoinak for excellent technical assistance. Sequencing and DNAmethylation analysis was performed by the SNP&SEQ Technology Platform in Uppsala. The facility is part of the National Genomics Infrastructure (NGI) Sweden and Science for Life Laboratory. The SNP&SEQ Platform is also supported by the Swedish Research Council and the Knut and Alice Wallenberg Foundation.
References
Footnotes
Contributors MW-H, AB, ERA, JI-K, GET, JM and GN conceived the study. MK, GN and AB recruited the patients. AB, ERA and JI-K performed the experiments. AB, JI-K, ERA, GET, JM and MW-H analysed the data. A-CS oversaw the RNA sequencing and DNA methylation analyses. AB and MW-H wrote the manuscript. All authors critically evaluated the manuscript and participated in the editing until its final version.
Funding The study was supported by grants from the Swedish Research Council, the Stockholm County Council, the Karolinska Institute, the Swedish Rheumatism association and the King Gustaf the V:th 80-year foundation and a Merck Research Collaboration grant.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The study was approved by the Regional Ethics Board in Uppsala and the Regional Ethics Committee in Stockholm.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request.