Article Text

Abstract
Objective To describe systemic sclerosis (SSc) with myopathy in patients without classic SSc-specific and SSc-overlap autoantibodies (aAbs), referred to as seronegative scleromyositis.
Methods Twenty patients with seronegative scleromyositis diagnosed by expert opinion were analysed retrospectively for SSc features at myositis diagnosis and follow-up, and stratified based on HEp-2 nuclear patterns by indirect immunofluorescence (IIF) according to International Consensus of Autoantibody Patterns. Specificities were analysed by protein A−assisted immunoprecipitation. Myopathy was considered an organ involvement of SSc.
Results SSc sine scleroderma was a frequent presentation (45%) at myositis diagnosis. Myositis was the most common first non-Raynaud manifestation of SSc (55%). Lower oesophagal dysmotility was present in 10 of 11 (91%) investigated patients. At follow-up, 80% of the patients met the American College of Rheumatology/EULAR SSc classification criteria. Two-thirds of patients had a positive HEp-2 IIF nuclear pattern (all with titers ≥1/320), defining three novel scleromyositis subsets. First, antinuclear antibody (ANA)-negative scleromyositis was associated with interstitial lung disease (ILD) and renal crisis. Second, a speckled pattern uncovered multiple rare SSc-specific aAbs. Third, the nuclear dots pattern was associated with aAbs to survival of motor neuron (SMN) complex and a novel scleromyositis subset characteriszed by calcinosis but infrequent ILD and renal crisis.
Conclusions SSc skin involvement is often absent in early seronegative scleromyositis. ANA positivity, Raynaud phenomenon, SSc-type capillaroscopy and/or lower oesophagal dysmotility may be clues for scleromyositis. Using HEp-2 IIF patterns, three novel clinicoserological subsets of scleromyositis emerged, notably (1) ANA-negative, (2) ANA-positive with a speckled pattern and (3) ANA-positive with nuclear dots and anti-SMN aAbs.
- Polymyositis
- Scleroderma
- Systemic
- Autoimmune Diseases
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
INTRODUCTION
Meeting the classification criteria of both systemic sclerosis (SSc) and myositis has been proposed as a definition of an SSc-myositis overlap syndrome.1–3 In the elaboration of the 2013 American College of Rheumatology (ACR)/EULAR SSc classification criteria, myositis was not selected as an SSc feature and instead was considered an SSc mimicker.4 Nevertheless, myositis has been reported with varying frequency in all serological subsets of SSc.5–8 Defining muscle involvement in SSc as scleromyositis 9 puts emphasis on the concept that SSc may also present as a myositis with only mild SSc features.
Classic SSc-specific autoantibodies (aAbs) include anti-centromere, -topoisomerase I, -RNA polymerase III, -Th/To and -U3RNP, whereas SSc-overlap aAbs include anti-U1RNP, -PM-Scl and -Ku. These aAbs are associated with fluorescent antinuclear antibody (ANA) patterns by indirect immunofluorescence (IIF) assay using the International Consensus of Autoantibody Patterns (ICAP) classification.10 In contrast, scleromyositis without classic SSc-specific or SSc-overlap aAbs has not been thoroughly described. This serological subset consists of several rare aAbs associated with nuclear and cytoplasmic fluorescent patterns, including anti-RuvBL1/2,6 anti-U4/U6RNP,11 anti-U5RNP,12 anti-U11/12RNP aAbs,7 anti-eIF2b13 as well as yet to be identified novel aAbs.
The objective of this study was to describe the clinicoserological features of 20 patients with seronegative scleromyositis, that is, with no classic SSc-specific and SSc-overlap aAbs. Patient characteristics were analysed for the presence of ACR/EULAR myositis,14 SSc criteria15 and any non-ACR/EULAR SSc features. Serological analyses using both IIF HEp-2 nuclear patterns10 and protein A−assisted immunoprecipitation of homogenates of 35S-metabolically labelled cells were performed to assess for the presence of rare SSc-specific aAbs.
METHODS
Patients
We conducted a retrospective study of 20 patients with seronegative SSc with a diagnosis of scleromyositis and without SSc-specific and SSc-overlap aAbs. Given the current absence of a gold standard for the definition of scleromyositis, this diagnosis was made by expert opinion (consensus of ≥2 experts). These patients were identified in a cohort of 340 patients with autoimmune myositis (AIM) at the Centre Hospitalier de l’Université de Montréal (CHUM) and Hôpital du Sacré-Coeur de Montréal (HSCM) (Montréal, Québec, Canada) recruited between 1967 and 2019, as previously described.16–18
Study variables
A retrospective medical record review using a standardised protocol was performed to collect clinical data, laboratory and imaging investigations, and muscle biopsy findings, as described previously in detail.16 17 Objective oropharyngeal dysphagia was defined by an abnormal videofluoroscopic swallowing study and/or the need for percutaneous gastrojejunostomy. Presence of lower oesophagal dysmotility was defined by either manometry or evidence on chest CT of lower oesophagal dilatation. Axial myopathy was characterised by weakness of the paraspinal muscles with head drop and/or camptocormia as prominent clinical features. Perimysial, perivascular or endomysial inflammation, and evidence of perifascicular atrophy, rimmed vacuoles and capillary abnormalities in muscle biopsies were recorded. Abnormal SSc-type capillaroscopy was defined as previously published.19
Inspired by the sequential occurrence of lung, muscle and joint manifestations in the time course of antisynthetase syndrome,20 we subclassified scleromyositis patients in four subsets according to their presenting feature at myositis diagnosis:
Patients meeting the 2013 ACR/EULAR SSc classification criteria were referred to as having a definite SSc phenotype at myositis diagnosis.
For patients not meeting the SSc classification criteria, the presenting SSc features could either be
Serology
Baseline immunological studies were performed at CHUM and HSCM as previously described.17 18 ANAs were determined by IIF on HEp-2 cells and the threshold value for positivity was >1:160. An anti-ENA panel was used to detect aAbs to centromere protein B, topoisomerase I and U1RNP. A commercial line blot assay (Myositis Profile 3 or 4, Euroimmun AG, Luebeck, Germany) was used to detect SSc-overlap anti-PM-Scl and anti-Ku aAbs.
Sera from patients without SSc-specific and SSc-overlap aAbs were collected and sent to Mitogen Diagnostics Laboratory, University of Calgary, where aliquots were biobanked at −80°C until needed. Samples were tested by IIF on HEp-2 substrate (Inova Diagnostics, San Diego, CA, USA) and read by technologists with >15 years of experience. HEp-2 IIF patterns were classified according to the ICAP standardised nomenclature (www.anapatterns.org).10 Anti-centromere aAbs were identified based on a discrete speckled nuclear pattern (AC-3).10 aAbs against centromere autoantigens CENP-A and CENP-B, topoisomerase I, RNA polymerase III (RP11 and RP155), PM-Scl (PM75 and PM100), Ro52/TRIM21, PDGFR, Ku, Th/To, NOR90/hUBF and U3RNP (fibrillarin) were detected by an SSc profile line immunoassay (Euroimmun AG). Overall, classic SSc-specific aAbs detected included aAbs to centromere proteins, topoisomerase I, RNA polymerase III, Th/To and U3RNP (fibrillarin) whereas SSc-overlap aAbs included aAbs to U1RNP, PM-Scl (75 and 100) and Ku. In the presence of an unexplained nucleolar IIF pattern, testing for anti-Th/To aAbs was also done, as previously described.30 31
All available sera were analysed for aAbs by protein A−assisted immunoprecipitation using HeLa cell extracts, both for nucleic acid analysis (RNA silver stain) and for proteins (metabolically labelled with 35S-methionine) as described.16 17 aAbs to survival of motor neuron (SMN)/gemins complex were tested in sera from 19 of the 20 patients by immunoprecipitation of 35S-methionine metabolically labelled K562 cell extracts as described,32 33 and were also tested by addressable laser bead immunoassay (ALBIA) for additional corroboration using purified, full-length, recombinant human SMN protein (Enzo Biochem, Farmingdale, New York, USA) with methods as previously described34 and results expressed as median fluorescence units (MFU) with positivity defined as 3 SD above the mean of normal and unrelated disease controls (>900 MFU). Using line blot assay and protein A−assisted immunoprecipitation, none of the 20 patients had aAbs to synthetases.
Statistical analysis
Descriptive statistics were used to describe the cohort. Comparisons among subsets were made using χ² analysis, Fisher two-tailed exact test, where applicable, or the Mann-Whitney U test. Kaplan-Meier curves were constructed to estimate 1-year, 5-year and 10-year survivals of the cohort.
Ethic statements
The study was approved by the CHUM (reference number 2015–5607-CE14.238) and HSCM (2014–1042) ethics committees.
RESULTS
Identifying 20 patients with seronegative scleromyositis, that is, with no classic SSc-specific and SSc-overlap aAbs
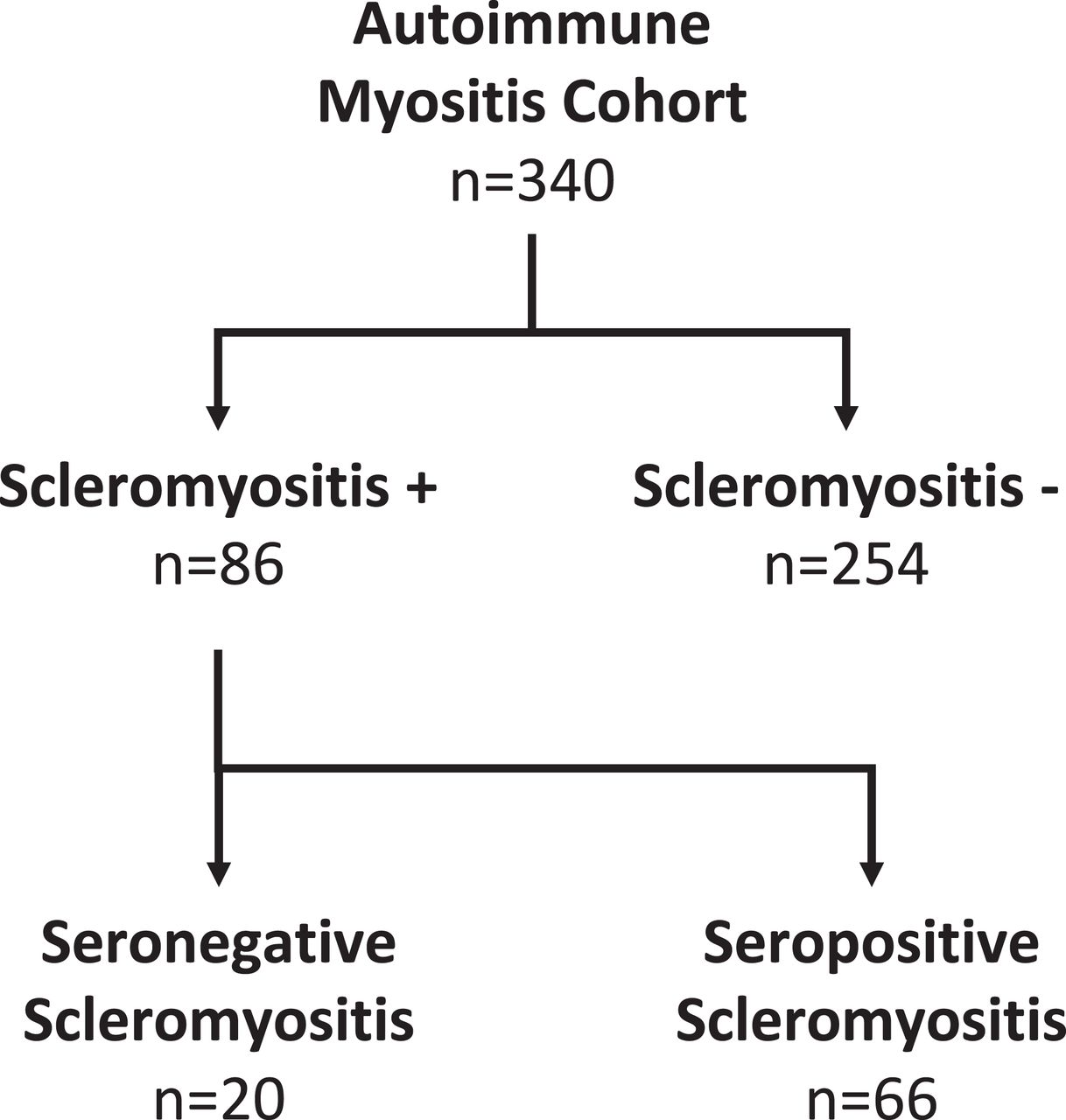
A diagnosis of scleromyositis was made by expert opinion in 86 of 340 patients with AIM of our cohort (figure 1), consisting of 19 patients with classic SSc-specific aAbs, 47 patients with SSc-overlap aAbs and 20 patients without classic SSc-specific and SSc-overlap aAbs. The latter group is referred to herein as seronegative scleromyositis and is the focus of this report. This group of patients included 3 men and 17 women, with a median age of 49.7 years (range 24.6–69 years) and a median follow-up of 6.5 years (range 3 months–32 years). One-year, 5-year and 10-year survivals were 90.0%, 78.5% and 56.6%, respectively. Cancer within 3 years of diagnosing myositis was identified in only one patient.
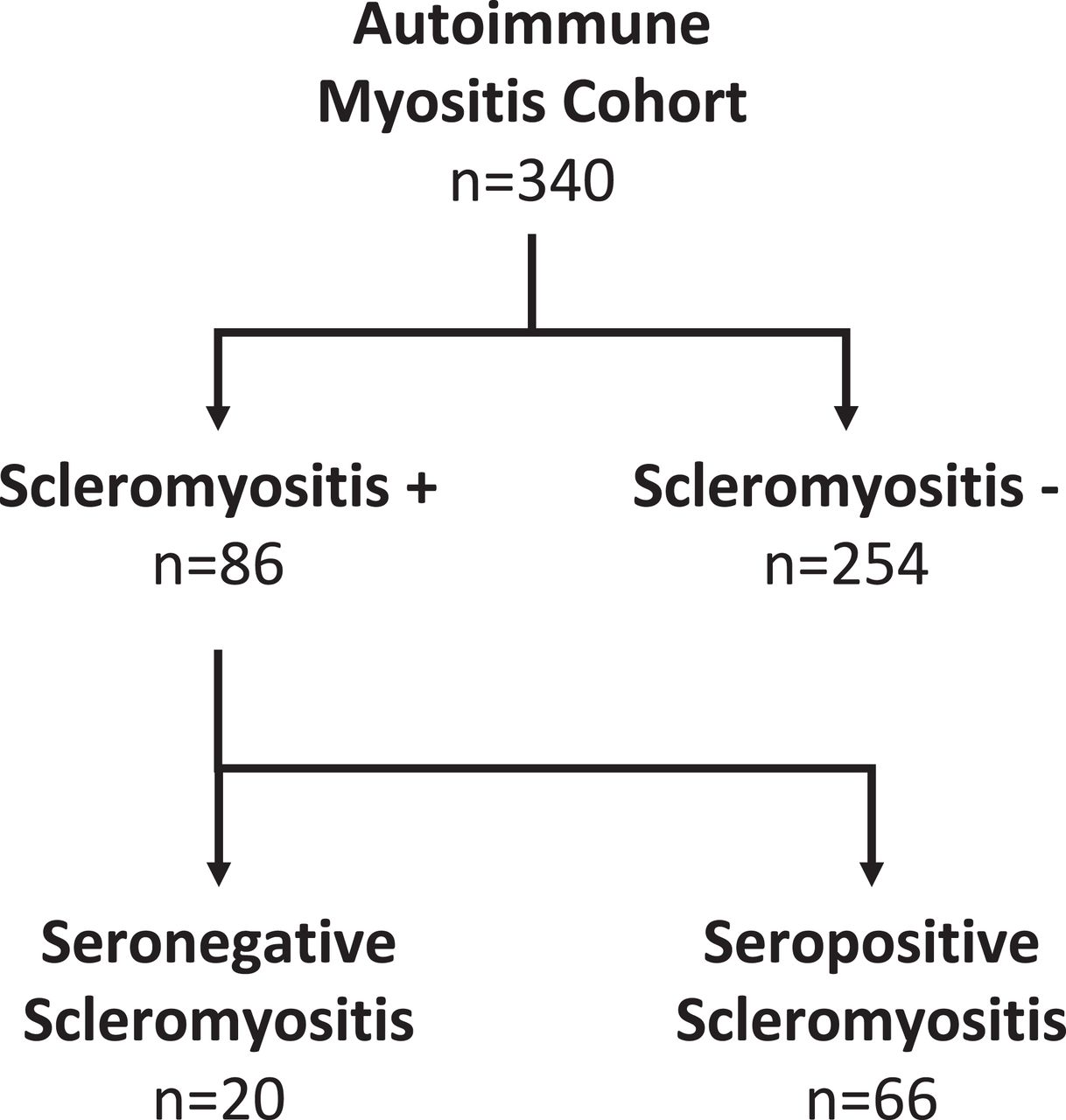
Identification of seronegative scleromyositis by expert opinion in an autoimmune myositis cohort.
Myopathic features in 20 patients with seronegative scleromyositis at myositis diagnosis
Proximal muscle weakness was documented in most patients (90%). Interestingly, one patient (patient 6, table 1) displayed a predominantly axial myopathy (camptocormia). Three patients (15%) presented with objective oropharyngeal dysphagia. Serum creatine kinase (CK) was elevated in all patients, with a median value of 1754 IU/L (range 300–12 410 IU/L). Myopathic electromyography (EMG) was observed in all 17 tested patients. Muscle biopsy was performed in all but two patients, and inflammation was documented in seven patients (perimysial and/or perivascular lymphocytic infiltrates in six patients and endomysial in one patient). Only 10 patients (50%) met the 2017 EULAR/ACR idiopathic inflammatory myopathy (IIM) classification criteria at myositis diagnosis. Probability of having IIM according to these criteria was (1) definite IIM in 10% (n=2/20), (2) probable IIM in 40% (n=8/20), (3) possible IIM in 0% and (4) not classifiable as IIM in 50% (n=10/20).
SSc features in 20 patients with seronegative scleromyositis at myositis diagnosis
At myositis diagnosis, only 11 patients (55%) met the 2013 ACR/EULAR SSc classification criteria (table 1). Definite SSc was therefore the presenting phenotype in these patients (n=11). Limited SSc skin involvement was observed in nine of them (82%). New-onset Raynaud phenomenon in the previous year was seen in 7 of these 11 patients (64%), whereas myositis was the first non-Raynaud SSc manifestation in 3 of them (27%).
SSc features at myositis diagnosis in 20 patients with seronegative scleromyositis
In the remaining patients (n=9/20) not fulfilling the 2013 ACR/EULAR SSc classification criteria, the presenting SSc features were either Raynaud phenomenon (n=5), ILD (n=1) or isolated SSc muscle involvement (n=3), respectively. All these patients presented without SSc skin involvement, that is, sine scleroderma, although three patients displayed telangiectasias and/or puffy fingers (table 1). Myositis was the most common first non-Raynaud SSc manifestation (55%), especially in patients presenting as sine scleroderma (n=8/9).
Taken together, SSc sine scleroderma (n=9/20, 45%) and limited cutaneous SSc (n=9/20, 45%) were the dominant subsets of SSc at myositis diagnosis, while diffuse cutaneous SSc (n=2, 10%) was less frequent. Interestingly, lower oesophagal dysmotility was present in 10 of 11 (91%) investigated patients, supporting an early diagnosis of SSc. Additionally, two-thirds (n=13/20) of patients with seronegative scleromyositis without classic SSc-specific and SSc-overlap aAbs had a positive HEp-2 IIF nuclear pattern (all with titers ≥1/320) (table 1).
Most patients with seronegative scleromyositis met the ACR/EULAR classification criteria for SSc at last follow-up
With the benefit of a median follow-up of 6.7 years (range 0.3–32 years), 80% (n=16/20) of patients with seronegative scleromyositis ultimately met the 2013 ACR/EULAR SSc classification criteria (table 2, online supplemental table 1).
Supplementary file 1
The SSc features leading to a diagnosis of SSc by expert opinion in the remaining four patients that did not meet the SSc classification criteria were as follows: Raynaud phenomenon (n=3), a speckled AC-4 or AC-5 IIF pattern (n=3), rare SSc-specific aAbs (anti-RuvBL1/2 and anti-U5RNP, n=2), lower oesophagal dysmotility (n=2), capillary damage on muscle biopsy (n=2), lower small-bowel dysmotility (n=2), abnormal nailfold capillaroscopy (n=1), isolated diffusing capacity of lung for carbon monoxide ≤70% of the normal predicted value (n=1), sclerodactyly (n=1) and telangiectasias (n=1).
SSc features at last follow-up in 20 patients with seronegative scleromyositis
Limited SSc (65%) was the dominant SSc subset at last follow-up, while diffuse SSc (20%) and SSc sine scleroderma (15%) were less frequent. In other words, six of nine patients (67%) initially presenting sine scleroderma ultimately developed an SSc skin involvement. At last follow-up, all patients but 1 (95%) had Raynaud phenomenon, whereas lower oesophagal dysmotility was a prominent feature encountered in 13 of 14 (93%) investigated patients. A single patient presented biopsy-proven myocarditis and developed pulmonary arterial hypertension.
Meeting the ACR/EULAR classification criteria for both myositis and SSc was uncommon at myositis diagnosis and last follow-up
At diagnosis of myositis, only two patients (10%) met both the 2017 EULAR/ACR IIM classification criteria and the 2013 ACR/EULAR SSc classification criteria (table 1). In contrast, at last follow-up, 40% (n=8/20) of the patients met both classification criteria.
HEp-2 IIF patterns uncover novel aAbs in seronegative scleromyositis
Sixty-five per cent of the patients with seronegative scleromyositis presented with a positive HEp-2 IIF (titers ≥1/320) while 35% were IIF-negative (table 1). When the patients were stratified according to their HEp-2 IIF status, three clinically relevant subsets emerged (figure 2 and online supplemental table 2).
Supplementary file 2
ANA-negative scleromyositis
All seven patients with HEp-2 IIF-negative scleromyositis had an abnormal SSc-type nailfold capillaroscopy, and ILD was present in five patients (71%). Most patients (71%) had serum CK ≤1000 IU/L and their CK levels were significantly lower than in patients with ANA-positive scleromyositis (p=0.039, online supplemental table 2). Patients wih ANA-negative scleromyositis were also at significantly higher risk for scleroderma renal crisis compared to ANA-positive scleromyositis patients (n=3/7 vs n=0/13, p=0.03).
ANA-positive scleromyositis
Speckled IIF patterns: sera from five patients with ANA-positive scleromyositis displayed a speckled nuclear pattern (AC-4 or AC-5 ICAP pattern). Interestingly, by immunoprecipitation, rare SSc-specific aAbs were found in four of these five (80%) patients: anti-RuvBL1/2 (n=2), anti-U4/U6RNP (n=1) and anti-U5RNP (n=1) (figure 2). At last follow-up, SSc small-bowel involvement (60%) and SSc sine scleroderma (40%) were key findings in these five patients.

HEp-2 nuclear patterns by indirect immunofluorescence assay and corresponding autoantibody specificities in seronegative scleromyositis. ANA, antinuclear antibody; SMN, survival of motor neuron.
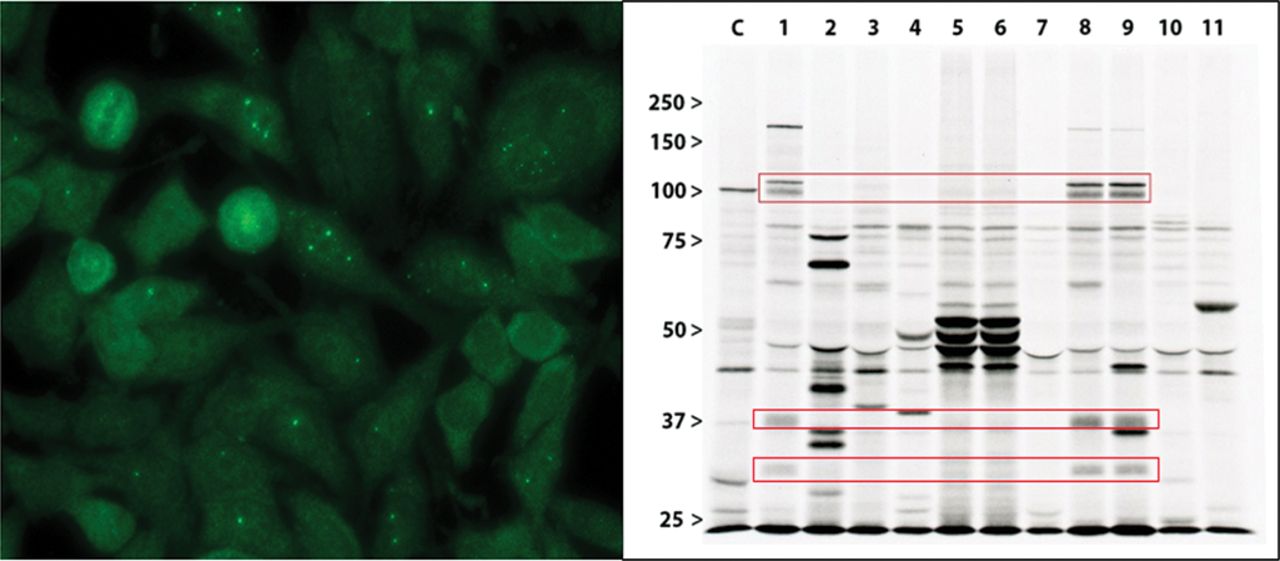
Nuclear dots IIF pattern: this pattern (AC-6 or AC-7 ICAP pattern) was observed in the sera from six patients with HEp-2 IIF-positive scleromyositis (figure 2 and online supplemental table 2). Importantly, five (83%) of these patients had anti-SMN aAbs by both immunoprecipitation and ALBIA (figure 3). Anti-SMN aAbs were not found by immunoprecipitation in other patients in this study. Of note, anti-SMN is typically associated with the few nuclear dots pattern (AC-7 pattern characterised by 1–6 dots) as shown in figure 3. However, not all sera had a clear-cut AC-7 pattern because some had >6 pleomorphic dots/nucleus than would be seen with classical AC-7. This may be explained by the presence of unidentified antigenic targets that give both an AC-6 and AC-7 pattern, as these sera were not monospecific when analysed (see figure 3).
The phenotype of patients with anti-SMN aAbs is shown in table 3. At diagnosis, three patients had limited cutaneous SSc, whereas two patients had SSc sine scleroderma. At last follow-up, four patients had limited SSc and a single patient had diffuse SSc. All patients were female with proximal weakness, displayed elevated serum CK levels (range 1494–3675 IU/L) and had an abnormal EMG. Arthritis and SSc calcinosis were each seen in three patients (60%), while digital ulcers, ILD, bilateral trigeminal neuropathy and small-bowel involvement with pneumatosis and retropneumoperitoneum were each seen in individual patients. No scleroderma renal crisis was observed at follow-up. Remarkably, one patient (patient 3, table 3) had three siblings suffering from spinal muscular atrophy.
{kind=link}
{kind=link}
{kind=link}
Anti-SMN autoantibodies are associated with few nuclear dots by indirect immunofluorescence on HEp-2 cells and react with the SMN complex by immunoprecipitation.
(A) Serum from patient 8 (table 3) with seronegative scleromyositis and anti-SMN autoantibodies by immunoprecipitation was associated by indirect immunofluorescence on HEp-2 cells with few nuclear dots pattern (AC-7 pattern according to the International Consensus on ANA patterns). (B) Immunoprecipitation using 35S-metabolically labelled K562 cell extracts was analysed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Lanes 1, 8 and 9 were respectively obtained with sera from seronegative scleromyositis patients 4, 13 and 14 (table 3). Bands at 100 kDa, 37 kDa and 30 kDa (red rectangles) represent multiple components of the SMN complex as determined with previously characterised index sera (19). Lane C, control serum with anti-PM-Scl autoantibodies. Lanes 5 and 6, consecutive sera from a patient with seronegative scleromyositis with anti-RUVBL1/2 autoantibodies. ANA, antinuclear antibody; SMN, survival of motor neuron.
Other HEp-2 IIF patterns: one patient had a nucleolar pattern (AC-8), while another patient had a homogeneous pattern (AC-1).
Phenotype of a novel scleromyositis subset associated with anti-SMN autoantibodies
DISCUSSION
There is a paucity of evidence on scleromyositis without classic SSc-specific and SSc-overlap aAbs. Only 10 such patients were included in a recent clinico-sero-pathological study of 37 patients with scleromyositis.25 In the present study, we describe in-depth 20 patients with seronegative scleromyositis that were identified from a cohort of 340 carefully phenotyped patients with AIM.16–18
SSc skin involvement is often absent in early scleromyositis
Knowledge of SSc features included in the 2013 ACR/EULAR SSc classification criteria is a step towards the early identification of SSc in patients with suspected AIM. However, these criteria put emphasis on the presence of SSc skin involvement, yet this feature was initially absent in almost half (45%) of our scleromyositis patients. The present study therefore explores serological, myological and non ACR/EULAR SSc features that led to an early diagnosis of scleromyositis by expert opinion.
Current definitions of scleromyositis are insensitive
In the absence of a gold standard defining scleromyositis, meeting the classification criteria of both IIM and SSc is often used in practice. However, defining scleromyositis as the fulfilment of these classification criteria lacks sensitivity, as only two patients (10%) in this study met this definition at myositis diagnosis. Interestingly, myositis was the first non-Raynaud manifestation of SSc in 11 patients (55%), also making the case for clinicians to recognise myositis as a potential feature of early SSc.
SSc sine scleroderma as a clue to early identification of scleromyositis in seronegative patients
Only 55% of our patients presented SSc skin involvement at myositis diagnosis, and the remaining patients presented with SSc sine scleroderma. Interestingly, most of them (89%) presented, singly or in combination, high titer HEp-2 IIF positivity, new onset Raynaud phenomenon, SSc-type capillaroscopy and/or lower oesophagal dysmotility, which are thus important clues to the early identification of scleromyositis in seronegative patients.
Interestingly, SSc sine scleroderma is a common presentation in patients with anti-Th-/To-positive SSc22 and, as shown herein, appears as a common presentation of seronegative scleromyositis. With the benefit of a median follow-up of 6.7 years, five of nine additional patients (total n=16/20, 80%) ultimately met the 2013 ACR/EULAR SSc classification criteria.
Using HEp-2 IIF patterns to subset clinically scleromyositis
When HEp-2 IIF patterns are analysed in seronegative scleromyositis, three clinically relevant subsets emerge: ANA-negative scleromyositis, ANA-positive scleromyositis with a speckled nuclear pattern (AC-4 or AC-5) and ANA-positive scleromyositis with nuclear dots (AC-6 or AC-7).
Although uncommon, HEp-2 IIF-negative scleromyositis was seen in 33% of the patients in this study. All patients had an abnormal nailfold capillaroscopy, ILD was frequent and, importantly, these patients were at high risk for scleroderma renal crisis, a feared complication of corticosteroid therapy in SSc,35 36 yet considered standard therapy in the treatment of myositis. Indeed, in the present study, two of the three HEp-2 IIF-negative patients who developed scleroderma renal crisis did so while treated with corticosteroids for their myositis.
HEp-2 IIF-positive scleromyositis with a speckled pattern (AC-4 or AC-5) was seen in 25% of the patients and was frequently associated with rarer SSc-specific aAbs, including anti-RuvBL1/2, anti-U4/U6RNP and anti-U5RNP. Of note, SSc sine scleroderma was the presenting phenotype in 60% of these patients. Although not as suggestive of SSc as a nucleolar pattern, a speckled pattern by IIF is a frequent serological finding in SSc when aAbs other than anti-centromere, anti-topoisomerase I and anti-RNA polymerase III are considered.37
Scleromyositis with nuclear dots on HEp-2 IIF (AC-6 or AC-7) was seen in 30% of our patients, and unearthed anti-SMN aAbs in five of six (83%) patients with this IIF pattern. Anti-SMN aAbs, all associated with nuclear dots on IIF, were originally described in three women with myositis (n=1) and myositis/SSc overlap syndrome (n=2)32 and, in a subsequent report, in a single patient with necrotising myopathy.33 Our data therefore suggest that the nuclear dots pattern may be used as a screening test for identifying anti-SMN aAbs. At last follow-up, all our patients with anti-SMN met the 2013 ACR/EULAR SSc classification criteria. Anti-SMN aAbs may therefore be a novel SSc-specific aAb.
Unique myopathological findings of myositis in SSc
Histopathological studies of muscle biopsies in SSc have suggested that myositis should be considered as a distinct feature of SSc23 24 38 39 because of unique myopathological findings.25 The presence of microangiopathy was demonstrated in scleromyositis associated with anti-centromere and anti-topoisomerase I aAbs.40 Necrotising myopathy and nonspecific myositis were the most common histopathological categories observed in SSc with muscle involvement.29 Also, fibrosing myopathy25 and acute neurogenic atrophy29 were recently proposed to be suggestive myopathological SSc findings. The full spectrum of myopathological features of scleromyositis, including in patients with anti-SMN aAbs, remains to be characterised.
Limitations of this study include its retrospective nature and the selection of cases based on expert opinion, given the lack of a gold standard for the definition of scleromyositis. On the other hand, a major strength of this study is the detailed clinical and phenotypic description of previously undescribed seronegative scleromyositis. SSc sine scleroderma was a common presenting SSc phenotype at myositis diagnosis and myositis was commonly the first non-Raynaud manifestation. High titer ANA positivity, new onset Raynaud phenomenon, SSc-type capillaroscopy and/or lower oesophagal dysmotility may be clues to the early identification of seronegative scleromyositis. HEp-2 IIF patterns allowed three novel clinical scleromyositis subsets to emerge. In particular, nuclear dots unearthed scleromyositis associated with anti-SMN aAbs, SSc calcinosis, low incidence of ILD and no scleroderma renal crisis.
Key messages
What is already known about this subject?
Scleromyositis is an emerging, yet poorly characterised, subset of myositis associated with features of systemic sclerosis.
What does this study add?
SSc skin involvement is often absent in early seronegative scleromyositis.
A nuclear dot HEp-2 IIF pattern is associated with anti-SMN autoantibodies and a novel scleromyositis subset.
How might this impact on clinical practice?
ANA positivity, Raynaud phenomenon, SSc-type capillaroscopy and/or lower oesophageal dysmotility are clues for early seronegative scleromyositis identification.
HEp-2 IIF patterns allow novel clinical scleromyositis subsets to emerge.
Acknowledgments
The authors thank Gemma Pérez, Haiyan Hou, Mei Feng Zhang and Tomoko Hasegawa for laboratory assistance.
REFERENCES
Footnotes
Contributors Conceptualisation and design: OL-C, AB-D, MS, MJF, YT and J-LS. Acquisition of data: OL-C, AB-D, SH, JB-T, A-MM, FZ, J-PM, JN, ER, J-RG, TG, MK, FJ, IR, IT, MS, MJF, YT and J-LS. Analysis and interpretation: OL-C, AB-D, SH, AM, VL, JB-T, MK, MH, MS, MJF, YT and J-LS. Critical revision of the manuscript for important intellectual content: all authors.
Funding This research was supported in part by the University of Montreal Scleroderma Research Chair (J-LS); by grants from Canadian Institutes of Health Research (J-LS, MK) (#MOP-142211), Sclérodermie Québec, Scleroderma Society of Ontario, Scleroderma Society of Canada, the Scleroderma Association of Saskatchewan, Scleroderma Manitoba, and the Scleroderma Association of British Columbia (J-LS, MK, SH, OL-C); by donations from Mrs Gisèle Sarrazin-Locas in support of the Autoimmunity Research Laboratory; and by a Japan Society for the Promotion of Science KAKENHI Grant-in-Aid for Scientific Research #15K08790 (MS). Dr Senécal holds the University of Montreal Scleroderma Research Chair.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.