Article Text

Abstract
Objectives To explore 6-month and 12-month secukinumab effectiveness in patients with axial spondyloarthritis (axSpA) overall, as well as across (1) number of previous biologic/targeted synthetic disease-modifying antirheumatic drugs (b/tsDMARDs), (2) time since diagnosis and (3) different European registries.
Methods Real-life data from 13 European registries participating in the European Spondyloarthritis Research Collaboration Network were pooled. Kaplan-Meier with log-rank test, Cox regression, χ² and logistic regression analyses were performed to assess 6-month and 12-month secukinumab retention, inactive disease/low-disease-activity states (Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) <2/<4, Ankylosing Spondylitis Disease Activity Score (ASDAS) <1.3/<2.1) and response rates (BASDAI50, Assessment of Spondyloarthritis International Society (ASAS) 20/40, ASDAS clinically important improvement (ASDAS-CII) and ASDAS major improvement (ASDAS-MI)).
Results We included 1860 patients initiating secukinumab as part of routine care. Overall 6-month/12-month secukinumab retention rates were 82%/72%, with significant (p<0.001) differences between the registries (6-month: 70–93%, 12-month: 53–86%) and across number of previous b/tsDMARDs (b/tsDMARD-naïve: 90%/84%, 1 prior b/tsDMARD: 83%/73%, ≥2 prior b/tsDMARDs: 78%/66%). Overall 6-month/12-month BASDAI<4 were observed in 51%/51%, ASDAS<1.3 in 9%/11%, BASDAI50 in 53%/47%, ASAS40 in 28%/22%, ASDAS-CII in 49%/46% and ASDAS-MI in 25%/26% of the patients. All rates differed significantly across number of previous b/tsDMARDs, were numerically higher for b/tsDMARD-naïve patients and varied significantly across registries. Overall, time since diagnosis was not associated with secukinumab effectiveness.
Conclusions In this study of 1860 patients from 13 European countries, we present the first comprehensive real-life data on effectiveness of secukinumab in patients with axSpA. Overall, secukinumab retention rates after 6 and 12 months of treatment were high. Secukinumab effectiveness was consistently better for bionaïve patients, independent of time since diagnosis and differed across the European countries.
- Spondyloarthritis
- DMARDs (biologic)
- Outcomes research
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
INTRODUCTION
Axial spondyloarthritis (axSpA) is a chronic, inflammatory, rheumatic disease characterised by inflammation and damage in the sacroiliac joints and spine, causing inflammatory back pain, disability and impaired quality of life.1 2 In patients with persistently high disease activity despite conventional treatment, biologic disease-modifying antirheumatic drugs (bDMARDs) are used, most often tumour necrosis factor inhibitors (TNFi).3
In 2015, the new-mode-of-action drug secukinumab was approved by the European Medicines Agency for use in ankylosing spondylitis. This fully human IgG1 monoclonal antibody targeting interleukin-17A4 has shown significant efficacy in the treatment of axSpA in randomised controlled trials (RCTs).5–7 According to current Assessment of Spondyloarthritis International Society (ASAS)-EULAR recommendations, secukinumab is recommended after failure of the first TNFi.3
There is to date limited real-world evidence on secukinumab treatment outcomes in patients with axSpA.8–10 Thus, the effectiveness of secukinumab, the impact of previous bDMARD or targeted synthetic DMARD (tsDMARD) use on secukinumab effectiveness in routine care, as well as the impact of time since diagnosis have not been studied in a large observational cohort of patients with axSpA.
Hence, the primary aim of this study was to determine the overall secukinumab retention rate in Europe after 12 months of treatment. Secondary aims were to determine 6-month retention rates and 6-month and 12-month inactive disease, low-disease-activity (LDA) and response rates. Primary and secondary aims were assessed overall as well as compared across number of prior b/tsDMARDs, across time since diagnosis and across European registries.
METHODS
European Spondyloarthritis Research Collaboration Network
The European Spondyloarthritis Research Collaboration Network (EuroSpA)11 was initiated in 2016/2017. EuroSpA aims to develop and investigate research questions by secondary use of prospectively collected real-life data on patients with SpA.11–13 So far, the collaboration includes 16 European registries, some of which have been collecting data from as early as 1999. Research questions focus on routine care treatment of patients with spondyloarthritis (SpA) in a European context, through pooling of relevant variables from the individual registries. All data are anonymised in the individual registries before upload through a secure virtual private network pipeline to the secured EuroSpA server, where the data are quality checked and pooled before statistical analyses are conducted.
Patients
For this study, anonymised data from patients with axSpA treated with secukinumab in routine care from 13 countries in the EuroSpA were uploaded and pooled: ARTIS (Sweden), RRBR (Romania), SCQM (Switzerland), ATTRA (Czech Republic), DANBIO (Denmark), BIOBADASER (Spain), TURKBIO (Turkey), NOR-DMARD (Norway), biorx.si (Slovenia), Reuma.pt (Portugal), GISEA (Italy), ROB-FIN (Finland) and ICEBIO (Iceland) (ordered from highest to lowest number of included patients). The data were collected independently of this study, that is, by national quality registries that collect information on any b/tsDMARDs. All patients were prospectively followed in the different registries, although the study was retrospectively designed with secondary use of data already collected in the registries, that is, planned after data collection had taken place, but before data were available. To be included patients had to be ≥18 years old, have a diagnosis of axSpA as judged by the treating rheumatologist, be previously secukinumab naïve and have a registered start date of secukinumab.
Assessments
Assessments included demographics, time since diagnosis, start and stop dates of secukinumab treatment, previous b/tsDMARD treatment (including the TNFis adalimumab, etanercept, infliximab, golimumab and certolizumab, the costimulatory inhibitor abatacept, the anti-B-cell agent rituximab, the interleukin (IL)-12/IL-23 inhibitor ustekinumab, the IL-6 receptor inhibitor tocilizumab, the IL-1 receptor inhibitor anakinra and the tsDMARDs apremilast, baricitinib and tofacitinib), current smoking status (yes/no), body mass index (kg/m2), C reactive protein (CRP, mg/L), erythrocyte sedimentation rate (mm/h), visual analogue scales (0–100 mm) of patient’s and evaluator’s global assessments, pain and fatigue (except for SCQM, biorx.si and RRBR which used a 0–10 numeric rating scale) and Health Assessment Questionnaire (0–3). The following composite indices were calculated based on their individual items: Bath Ankylosing Spondylitis Disease Activity Index (BASDAI, 0–10),14 Bath Ankylosing Spondylitis Functional Index (BASFI, 0–10)15 and Ankylosing Spondylitis Disease Activity Score (ASDAS; including BASDAI questions 2, 3 and 6, patient’s global assessment and CRP).16 17 The following inactive disease, LDA and response measures were calculated at 6-month and 12-month follow-up: BASDAI remission defined as <2, BASDAI LDA defined as <4,18 ASDAS inactive disease (<1.3),19 ASDAS LDA (<2.1),19 change in BASDAI and ASDAS, BASDAI50 response (at least 50% improvement in BASDAI score or an absolute change of 2 units on a 0–10 scale),18 ASAS20/40 response (including measures of physical function, pain, inflammation as assessed by duration of morning stiffness and patient’s global assessment),20 21 ASDAS clinically important improvement (ASDAS-CII≥1.1) and ASDAS major improvement (ASDAS-MI≥2.0).22
Primary and secondary outcomes
The primary outcome was the overall 12-month secukinumab retention rate. Secondary outcomes were the overall 6-month secukinumab retention rate, and 6-month and 12-month inactive disease, LDA and response rates. Primary and secondary outcomes were compared across (1) number of previous b/tsDMARDs (0/1/≥2), (2) time since diagnosis (<2/2–4/>4 years) and (3) the different European registries.
Statistical analyses
Statistical analyses were performed according to a predefined statistical analysis plan. Normally distributed data were presented as mean (SD) and skewed data as median (IQR). Group comparisons of demographics and baseline disease activity were performed with analysis of variance (ANOVA), Kruskal-Wallis or χ² test, as appropriate.
All drug retention, disease state and response analyses were performed at 6-month and 12-month follow-up. Drug retention was explored by Kaplan-Meier analyses with log-rank test. Cox regression analyses adjusted for age, gender and time since diagnosis were applied for comparisons of retention across number of previous b/tsDMARDs (0/1/≥2) and across European registries, and adjusted for age and gender for comparisons across time since diagnosis (<2/2–4/>4 years).
Group comparisons of disease states, changes in BASDAI/ASDAS and response rates were performed with ANOVA, Kruskal-Wallis or χ² test, as well as with analysis of covariance (ANCOVA) and logistic regression analyses adjusted for age, gender and time since diagnosis (comparisons across number of previous b/tsDMARDs (0/1/≥2) and across European registries), or age and gender (comparisons across time since diagnosis (<2/2–4/>4 years)), as appropriate. LUNDEX23 adjustments (crude value adjusted for drug retention) were calculated for disease states.
Multivariate Imputation by Chained Equations (including 50 imputations) was performed for the Cox regression, ANCOVA and logistic regression analyses for 300 patients with missing data for time since diagnosis. The remaining analyses were available case analyses. Number of patients with available data for each of the analyses are shown in table 1 and in online supplemental tables 1–5.
Demographics and baseline disease activity measures for all patients, as well as compared across number of previous b/tsDMARDs
Supplemental material
Observations were censored by the date of data extraction, date of death or end of registry follow-up, whichever came first. The index date was defined as the secukinumab treatment start date. Follow-up period was defined as the period between index date and the end of the 12-month study period, date of death or end of registry follow-up/capture, whichever came first. A significance level of 0.05 was set. No corrections for multiple comparisons were performed. Statistical analyses were performed with R version 3.4.4/3.6.1 and SPSS version 25.
Ethics
The study was approved by the respective national data protection agencies and research ethical committees according to legal regulatory requirements in the participating countries. It was performed in accordance with the Declaration of Helsinki and followed the Strengthening the Reporting of Observational Studies in Epidemiology guidelines.24
RESULTS
A total of 1860 patients with axSpA were included, thereof 414 b/tsDMARD naïve patients, 448 patients previously treated with 1 b/tsDMARD and 998 patients previously treated with 2 or more b/tsDMARDs. The 1 prior b/tsDMARD group included <10 patients who had previously been treated with ustekinumab and rituximab and 444 patients previously treated with a TNFi. tsDMARDs had previously been used by 14 patients, all in the ≥2 prior b/tsDMARD group, including apremilast by 10 patients and tofacitinib and baricitinib by <10 patients; they had all previously also been treated with TNFi. The remaining 984 patients had previously exclusively been treated with different bDMARDs, including TNFi used by all of the patients, ustekinumab by 24 patients, abatacept by 27, rituximab by 12, tocilizumab by 28 and anakinra by <10 patients (table 1).
Information on the modified New York criteria were available in 664 patients and fulfilled in 514 of these patients, and information on the ASAS criteria in 963 patients and fulfilled in 799 of these patients. Furthermore, 97 patients were registered as fulfilling the ASAS criteria but not the modified New York criteria, that is, compatible with non-radiographic axSpA.
Patients were only included in the study if they had been followed in the individual registries since start of secukinumab treatment. Hence, date of secukinumab initiation had no missing cases. Of the 1860 patients included in the study, 916 patients were still using secukinumab at the date of data extraction, 659 patients had a registered stop date of secukinumab, 240 patients had a date of end of follow-up and the remaining 45 patients were assumed to have ended secukinumab 12 months after the last registered visit, in accordance with the predefined statistical analysis plan.
Information on doses was not registered systematically. Of 828 patients in whom doses were registered, 762 patients initiated secukinumab 150 mg/month and 66 patients 300 mg/month.
The patients started secukinumab treatment between April 2015 and December 2018. Data cuts in the individual registries were performed between October 2018 and September 2019. A total of 1270 of the 1860 patients had started secukinumab treatment at least 52 weeks prior to the date of the data cut. b/tsDMARD-naïve patients were younger, had shorter time since diagnosis, higher baseline disease activity and a higher proportion were men compared with patients treated with one prior, or two or more prior, b/tsDMARDs.
Secukinumab drug retention overall and compared by previous b/tsDMARD treatment
Overall, 6-month and 12-month secukinumab retention rates were 82% and 72%, respectively, and differed significantly (p≤0.001) across number of previous b/tsDMARDs, with decreasing drug retention rates with increasing previous b/tsDMARD use (6-/12-month: b/tsDMARD naïve: 90%/84%, 1 prior b/tsDMARD: 83%/73%, ≥2 prior b/tsDMARDs: 78%/66%; table 2, figure 1).
Treatment effectiveness after 6 and 12 months of secukinumab treatment
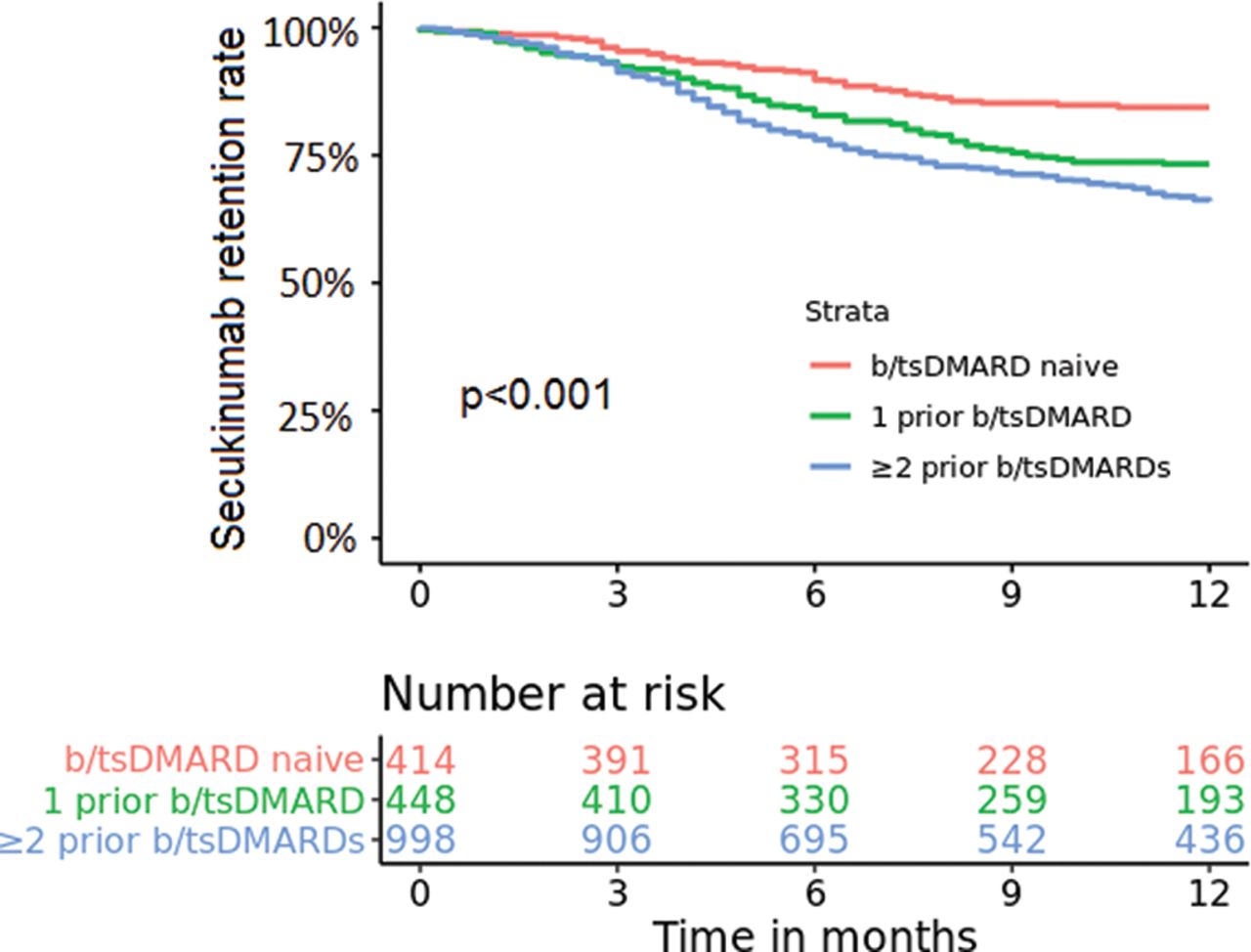
Pooled 12-month secukinumab retention rates for patients with axial spondyloarthritis in the European Spondyloarthritis Research Collaboration Network stratified by previous biologic/targeted synthetic disease-modifying antirheumatic drug (b/tsDMARD) treatment (log-rank test; p<0.001).
When adjusted for age, gender and time since diagnosis, patients who had used one previous b/tsDMARD or two or more previous b/tsDMARDs were at higher risk of discontinuing secukinumab before 12 months of treatment compared with b/tsDMARD-naïve patients (HR 1.78, 95% CI 1.29 to 2.47 and HR 2.33, 95% CI 1.74 to 3.11, respectively; online supplemental figure 1).
Secukinumab retention according to reason for withdrawal
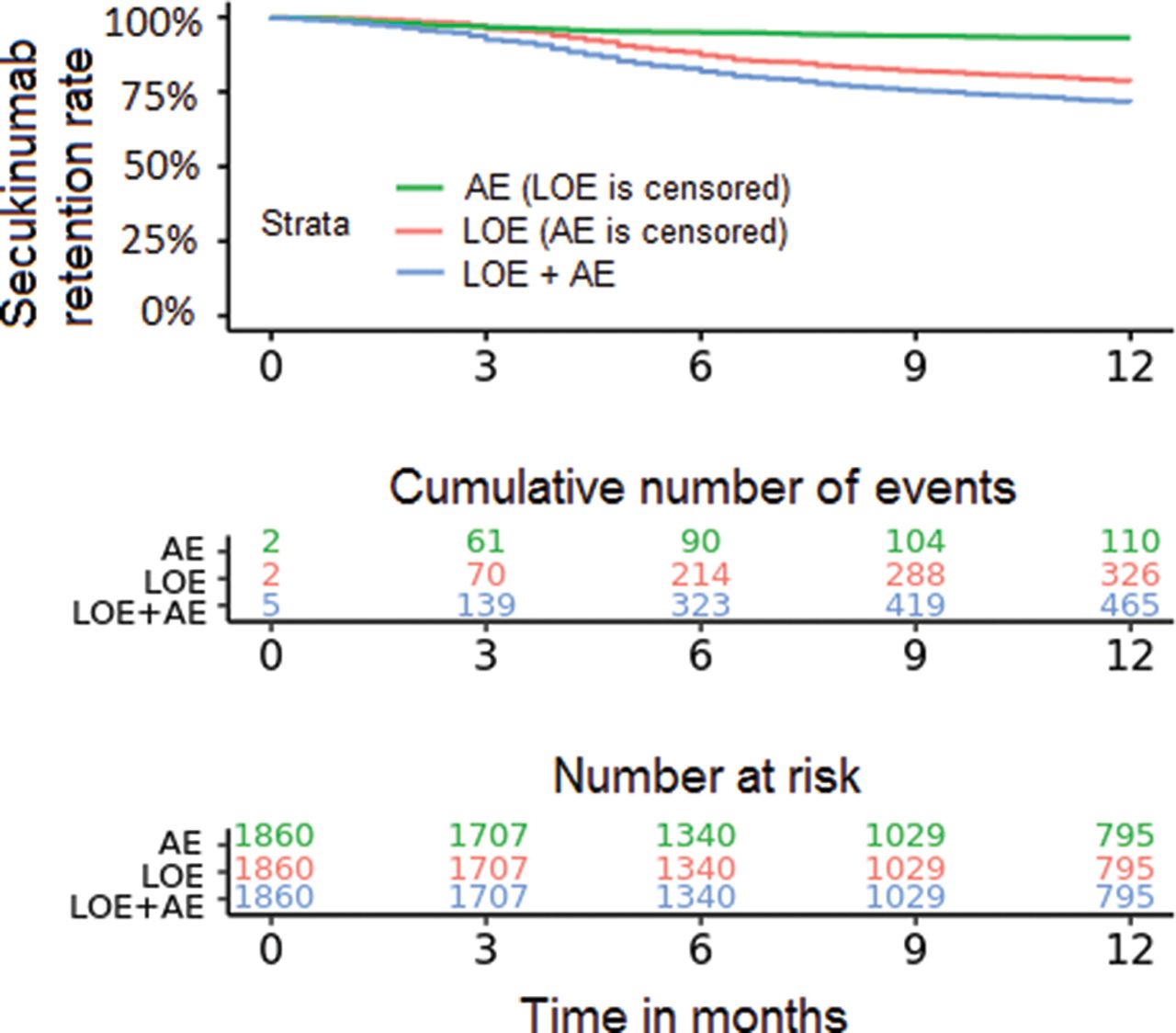
More patients withdrew from secukinumab due to loss of efficacy (n=326) than adverse events (n=110) (figure 2).
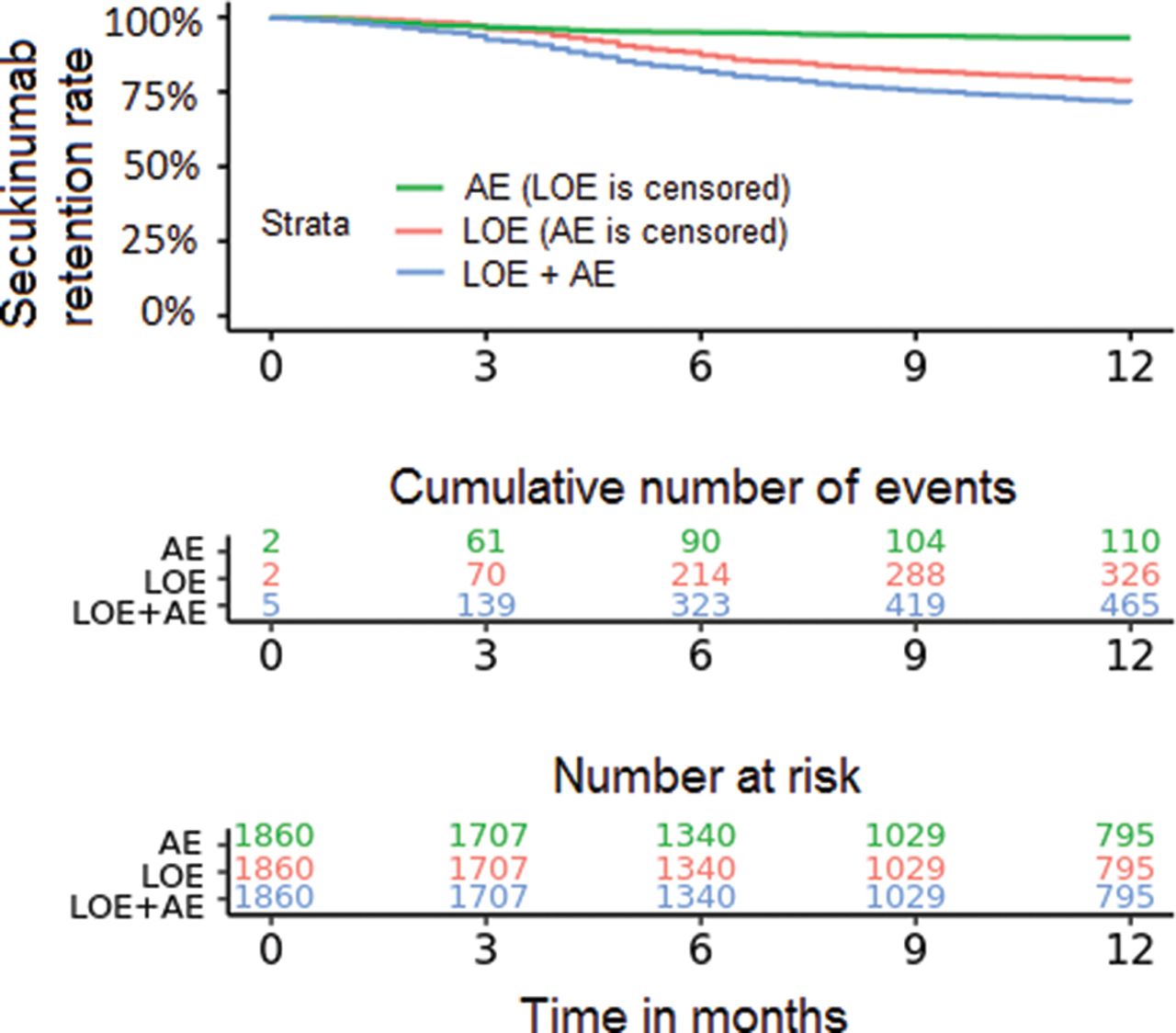
Secukinumab retention rates due to adverse events (AE) and loss of efficacy (LOE, Kaplan-Meier plot; for 29 patients, it was not distinguished by the registries whether reason for withdrawal was due to AE or LOE).
Secukinumab retention rates according to time since diagnosis
Drug retention of secukinumab was not associated with time since diagnosis, when patients were stratified into time <2 years, 2–4 years and >4 years since diagnosis (online supplemental table 3). Additional adjustment for age and gender gave similar findings (6 months, p=0.83; 12 months, p=0.85).
Retention, inactive disease, LDA and response rates after 6 and 12 months of secukinumab treatment stratified across European registries (ICEBIO (Iceland) is excluded from the table due to <10 patients at secukinumab initiation)
Secukinumab retention rates across European countries
Significant heterogeneity in baseline demographics, disease activity measures and proportions of bionaïve patients across different European countries were found (online supplemental table 4). Secukinumab retention rates after 6 and 12 months of treatment varied significantly across the countries in EuroSpA (table 3, figure 3). Adjustment for age, gender and time since diagnosis gave similar findings (data not shown).

Twelve-month secukinumab retention rates compared across patients with axial spondyloarthritis in different European registries (Kaplan-Meier with log-rank test; ICEBIO (Iceland) and 12-month number at risk for TURKBIO (Turkey) are excluded from the plot due to <10 patients).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Bar charts of proportions of patients achieving different disease state and response rates after 6 and 12 months of secukinumab treatment compared across previous biologic/targeted synthetic disease-modifying anti-rheumatic drug (b/tsDMARD) treatment. ASAS20/40, Assessment of Spondyloarthritis International Society 20/40 response; ASDAS CII, ASDAS clinically important improvement (≥1.1); ASDAS MI, ASDAS major improvement (≥2.0); BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; BASDAI50, 50% improvement in BASDAI.
Inactive disease, LDA and response rates after 6 and 12 months of secukinumab treatment
Median BASDAI after 6 and 12 months of secukinumab treatment were 3.9 and 3.9, and ASDAS 2.6 and 2.5, respectively (table 2). Crude/LUNDEX-adjusted BASDAI<2 was achieved by 26%/21% of the patients after 6 months and 25%/16% after 12 months, and BASDAI<4 by 51%/40% of the patients after 6 months and 51%/32% after 12 months of treatment. Proportions of patients achieving crude/LUNDEX-adjusted ASDAS inactive disease after 6 and 12 months of treatment were 9%/7% and 11%/7%, and ASDAS low disease activity 24%/19% and 27%/17%, respectively. After 6 and 12 months of treatment, BASDAI50 response was achieved by 53% and 47%, ASAS20 response by 40% and 37%, ASAS40 response by 28% and 22%, ASDAS-CII by 49% and 46% and ASDAS-MI by 25% and 26% of the patients, respectively (table 2). Response rates in patients having received ≥3 b/tsDMARDs can be found in online supplemental table 5 and were overall comparable to the group of patients who had previously used ≥2 b/tsDMARDs. There were some differences in baseline characteristics between patients with missing and non-missing data for 6-month and 12-month response rates, but these were not consistent across the different response criteria (data not shown).
Disease states and response rates across number of previous b/tsDMARDs
Crude and LUNDEX-adjusted inactive disease, LDA and response rates after 6 and 12 months of treatment varied statistically significantly across number of previous b/tsDMARDs (all p≤0.002), showing decreasing effectiveness with increasing previous b/tsDMARD use. Overall, the numerically highest rates were found in bionaïve patients (table 2, figure 4). Adjustment for age, gender and time since diagnosis gave similar results (data not shown).
Inactive disease, LDA and response rates according to time since diagnosis
Disease states and response rates at 6 and 12 months were not significantly different between patients with time since diagnosis <2 years, 2–4 years and >4 years, except for achievement of BASDAI<2 and <4 at 6 months (online supplemental table 3). Adjustment for age and gender gave similar results.
Disease states and response rates across the European registries
Disease states and response rates after 6 and 12 months of treatment varied significantly across the registries in EuroSpA, except for ASDAS inactive disease (table 3). Adjustment for age, gender and time since diagnosis did not change this (data not shown).
DISCUSSION
In this study of 1860 patients from 13 European countries, we present the first comprehensive real-life data on effectiveness of secukinumab in patients with axSpA. Overall, secukinumab retention rates after 6 and 12 months of treatment were high (82% and 72%, respectively). Importantly, effectiveness differed significantly across number of previous b/tsDMARDs, with bio-naïve patients consistently having numerically better secukinumab retention as well as disease state and response rates. Time since diagnosis did not impact on secukinumab drug retention, inactive disease/LDA or response rates, with few exceptions for 6-month BASDAI outcomes. Significant differences between the European registries were found.
To date, only a few small observational studies on secukinumab effectiveness in axSpA have been published, including a UK study of 76 patients that reported a trend towards improved 6-month BASDAI and BASFI responses,10 an Italian study of 39 patients with axSpA that reported good 2-year effectiveness of secukinumab9 and a German pilot study of 13 patients that reported 2-year clinical improvement and regression of spinal inflammation as assessed by MRI.8 Therefore, this real-life, international multicentre study from large observational cohorts represents an important addition to the RCTs on secukinumab. Interestingly, data on secukinumab retention from RCTs are scarce. In the MEASURE 1 trial, the 2-year secukinumab retention was 78%,25 and in the MEASURE 2 trial, the 3-year retention rate was 86% for secukinumab 150 mg every 4 weeks.6 In the MEASURE 3 trial, the 1-year retention rate was 87% in the group of patients who were initially randomised to 150 mg secukinumab,7 which is higher than the overall 1-year retention rate of 72% in this real-life study. Compared to the patients in the MEASURE 2 and 3 trials, the patients in our study were older (mean age 47 vs 42 and 43 years), had lower baseline BASDAI (mean BASDAI 6.0 vs 6.6 and 7.0), longer time since diagnosis (9.9 vs 7.0 and 6.0 years), a lower proportion were TNFi-naïve (22% vs 61% and 57%) and we included both patients with radiographic axSpA and patients with non-radiographic axSpA, whereas only patients with radiographic axSpA were included in the MEASURE 2 and 3 trials.6 7 Due to these differences in patient characteristics and, perhaps most importantly in study design, no valid conclusion on the comparison of secukinumab effectiveness across these studies may be drawn.
The 12-month secukinumab retention rate for the b/tsDMARD-naïve patients in this study (84%) was similar to the recently reported 12-month retention rate to first-line TNFi in patients with axSpA in Europe (80%)11 as well as to other studies on first-line TNFi treatment.26 Retention to a first TNFi in axSpA is in general known to be higher than to a second or third TNFi.27 Of note, we report similar findings for secukinumab in this study, with numerically higher secukinumab retention for b/tsDMARD-naïve patients compared with patients treated with one and two or more previous b/tsDMARDs. Thus, this study supports the effectiveness of using secukinumab as first-line bDMARD in the routine management of axSpA.
For b/tsDMARD-naïve patients, achievement of 12-month BASDAI<4 (76%) and ASAS20/40 response (69%/55%) in the current study were comparable to first-time TNFi-treated patients with axSpA in a recently published large-scale European study, in which 12-month BASDAI<4 was achieved by 75% and ASAS20/40 by 67%/53% of the patients, respectively.11 In the same study, ASDAS inactive disease was achieved by a higher proportion of patients than in the current study (35% vs 18%). It should be emphasised, however, that several parameters, including time since diagnosis and baseline disease activity (ASDAS and BASDAI), were markedly higher in the secukinumab-treated patients than in the TNFi-treated patients, limiting direct comparisons.
Compared with the MEASURE 2 trial, 12-month response rates for secukinumab were lower in our study including BASDAI50 (47% vs 51%), mean change in BASDAI (−2.1 vs −3.2), ASAS20 (37% vs 74%) and ASAS40 response (22% vs 57%).6 These differences were less pronounced for b/tsDMARD-naïve patients (for ASAS20, 69% in the present study vs 80% in the MEASURE 2 trial and for ASAS40, 55% vs 63%).6 In the MEASURE 3 trial, ASAS20/40 response rates at week 52 were 54%/41%, respectively, for the 150 mg secukinumab group.7
Of note, comparison of treatment outcomes across different studies is challenging due to heterogeneity in study populations and inclusion criteria and valid conclusions may usually not be drawn. Direct comparison of the efficacy of IL-17 pathway inhibition and TNFi in a head-to-head design as first-line b/tsDMARD treatment in axSpA is warranted28 29 as well as more observational studies on the long-term effectiveness of IL-17 pathway inhibition.
Fewer patients withdrew from secukinumab due to adverse events than lack of efficacy. Whereas about 6% of the patients in our study withdrew from secukinumab up to week 52 due to adverse events, 4% of the patients in the initial 150 mg secukinumab group in the MEASURE 3 trial withdrew from secukinumab due to adverse events up to week 52.7
The major strengths of this study are the inclusion of large cohorts of patients from observational registries across Europe, reflecting routine care in a wide range of European countries, and the reporting of 12-month observational data on secukinumab. The limitations of the study are the inherent limitations of registry studies compared to RCTs, that is, a lower data quality than in RCTs, for example, missing data on response outcomes across the registries. The use of the BASDAI cut-offs <2 and <4 was chosen. There is no consensus as yet on the best cut-off for BASDAI remission in axSpA. However, these cut-offs were prespecified, a cut-off of 4 has previously been used for active disease,18 and we also assessed ASDAS inactive disease and ASDAS LDA in line with the current recommendations.2 Furthermore, we assessed several other response criteria specifically developed for axSpA.18 Heterogeneity in secukinumab effectiveness across the registries was seen. Importantly, the proportions of bionaïve patients varied considerably across registries and may explain some of the heterogeneity in effectiveness across the registries. Furthermore, number of included patients varied considerably across the registries, which may also challenge direct comparisons. Still, one of the major advantages of data pooling is the overall high number of patients, enabling to explore research questions often not feasible in a single register.
In conclusion, in this large-scale real-life study, the overall 12-month secukinumab retention was high, with differences between the European registries. Response and rates of favourable disease states were lower than for RCTs on secukinumab, but comparable to available real-world data on TNFi. Drug retention, favourable disease states and response rates differed significantly across the number of previous b/tsDMARDs, showing overall better outcomes with less previous use of b/tsDMARDs, whereas time since diagnosis had no impact on secukinumab effectiveness. The study supports the effectiveness of secukinumab in the treatment of axSpA, but also underlines the need for head-to-head studies on treatment effectiveness of TNFi and IL-17 pathway inhibitors.
Key messages
What is already known about this subject?
Secukinumab has in RCTs shown efficacy in the treatment of axSpA, but observational studies confirming these findings are lacking.
What does this study add?
Secukinumab retention, inactive disease, low-disease-activity and response rates differed significantly across number of previous b/tsDMARDs, with overall better outcomes with less previous use of b/tsDMARDs, whereas time since diagnosis had no significant impact on outcomes.
The overall 6- and 12-month secukinumab retention was high and comparable to studies on TNFi.
Response rates were lower than in RCTs, but post-treatment disease states and response rates were comparable to studies on TNFi.
How might this impact on clinical practice?
The study supports the effectiveness of secukinumab in the treatment of axSpA, but also underlines the need for head-to-head studies on treatment effectiveness of TNFi and IL-17 pathway inhibitors.
Acknowledgments
Thanks to Novartis Pharma AG and IQVIA for supporting the EuroSpA collaboration. Novartis had no influence on the data collection, statistical analyses, manuscript preparation or decision to submit.
REFERENCES
Footnotes
MLH and MØ contributed equally
Contributors The study protocol and analysis plan was drafted by BM, MØ and MLH and revised and approved by all authors. Data analyses were done by BM and draft of the manuscript by BM, MØ, MLH and LØ. All authors have contributed substantially to the acquisition and interpretation of data, revised the manuscript for important intellectual content and approved the final submitted version.
Funding The EuroSpA collaboration was financially supported by Novartis. Novartis had no influence on the data collection, statistical analyses, manuscript preparation or decision to submit. The national registries have received financial support from a range of pharmaceutical companies, including Novartis. These funds are given as unrestricted grants.
Competing interests BM: consultancy fees and research grant from Novartis. UL: None. CC: None. AC: fees for speaking and/or consulting from AbbVie, Celgene, Eli Lilly, Janssen-Cilag, Merck Sharp & Dohme, Novartis, Pfizer and UCB. JZ: none. AGL: research grant from Novartis, consultant for/served on the speakers bureau for AbbVie, MSD, Novartis, Pfizer, Roche, and UCB. MP-S: speaker and consultancy fees from Abbvie, BMS, Janssen, Lilly, MSD and Sanofi. FO: speaker and consultancy fees from Abbvie, Abdi Ibrahim, Amgen, Celltrion, Eli Lilly, Novartis, Pfizer, Roche and UCB and research grant from Pfizer. TKK: fees for speaking and/or consulting from AbbVie, Amgen, Biogen, Celltrion, Egis, Eli Lilly, Hikma, MSD, Mylan, Novartis/Sandoz, Oktal, Orion Pharma, Hospira/Pfizer, Roche, Sanofi and UCB and received research funding to Diakonhjemmet Hospital from AbbVie, BMS, MSD, Pfizer, Roche and UCB. ZR: speaker and consultancy fees from Abbvie, Amgen, Biogen, Eli Lilly, Medis, MSD, Novartis, Pfizer, Roche, Sanofi. MJS: speaker fees from Novartis and Pfizer. FI: speaker and consulting fees from AbbVie, Eli Lilly, Novartis, Pfizer, Roche, Sanofi, UCB, MSD. AMH: research grant from MSD. BG: Speaker fee from Novartis. JA: none. RI: consulting fees from Abbvie, Eli-Lilly, Ewopharma, Novartis, Pfizer, Roche, Sandoz. MJN: fees for speaking and/or consulting from AbbVie, Celgene, Eli Lilly, Novartis and Pfizer. KP: honoraria for lectures from MSD, BMS, AbbVie, UCB, Roche, Biogen, Amgen, Novartis, Pfizer, Medac, Egis. CS-P: none. SA: has received grant/research support from, been a consultant for, and served on the speakers bureau for AbbVie, MSD, Novartis, Pfizer, Roche, and UCB. JS: none. MT: none. HS: speaker fees from Novartis, Pfizer and Abbvie. MS: none. JÖ: none. AjG: None. GM: none. IvdH-B: Fees for speaking and/or consulting from Abbvie, Lilly, Novartis, BMS, MSD, UCB and Pfizer. SG: none. CHB: research grant from Novartis. LØ: research grant from Novartis. MH: grant from AbbVie, Biogen, BMS, Novartis, Pfizer, UCB. Personal fee from CellTrion, MSD, Orion, Samsung. MO: consultancy fees and/or speaker fees form Abbvie, BMS, Boehringer-Ingelheim, Celgene, Eli-Lilly, Hospira, Janssen, Merck, Novartis, Novo, Orion, Pfizer, Regeneron, Roche, Sandoz, Sanofi and UCB; and research support from Abbvie, BMS, Celgene, Merck, and Novartis.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplemental information.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.