Article Text

Abstract
Objectives Review of efficacy and safety of Janus kinase (JAK) inhibition in immune-mediated inflammatory diseases (IMIDs).
Methods A systematic literature research (SLR) of all publications on JAK inhibitors (JAKi) treatment published until March 2019 using MEDLINE, EMBASE and the Cochrane Library. Efficacy and safety were assessed in randomised controlled trials (RCTs), integrating long-term extension periods additionally for safety evaluation.
Results 3454 abstracts were screened with 85 included in the final analysis (efficacy and RCT safety: n=72; safety only: n=13). Efficacy of RCTs investigating tofacitinib (TOFA, n=27), baricitinib (BARI, n=9), upadacitinib (UPA, n=14), filgotinib (FILGO, n=7), decernotinib (DEC, n=3) and peficitinib (PEF, n=7) was evaluated. Six head-to-head trials comparing JAKi with tumour necrosis factor inhibitors (TNFi) were included. Efficacy of JAKi was shown in rheumatoid arthritis (RA) for all agents, psoriatic arthritis (TOFA, FILGO), ankylosing spondylitis (TOFA, FILGO), systemic lupus erythematosus (BARI), chronic plaque psoriasis (TOFA, BARI, PEF), ulcerative colitis (TOFA, UPA), Crohn’s disease (UPA, FILGO) and atopic dermatitis (TOFA, BARI, UPA). Safety analysis of 72 RCTs, one cohort study and 12 articles on long-term extension studies showed increased risks for infections, especially herpes zoster, serious infections and numerically higher rates of venous thromboembolic events. No increased malignancy rates or major adverse cardiac events were observed.
Conclusion JAKi provide good efficacy compared to placebo (and to TNFi in RA and Pso) across various IMIDs with an acceptable safety profile. This SLR informed the task force on points to consider for the treatment of IMIDs with JAKi with the available evidence.
- Arthritis
- rheumatoid
- psoriatic
- spondylitis
- ankylosing
- lupus erythematosus
- systemic
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
INTRODUCTION
The first randomised controlled trial (RCT) investigating the inhibition of Janus kinases (JAK) via the JAK-1/2 selective agent ruxolitinib/INCB018424 (RUXO) in patients with an immune-mediated inflammatory disease (IMID), namely rheumatoid arthritis (RA), was completed in 2008; however, this study has not been published until today (ClinicalTrials.gov identifier: NCT00550043). Since then, numerous trials on JAK inhibitors (JAKi) have been conducted in various IMIDs across many disciplines, including rheumatology, dermatology and gastroenterology.1–14
In 2012, tofacitinib (TOFA), a JAK-1/2/3 inhibitor, was the first agent to be approved for an IMID, namely RA, but subsequently also for treating paients with psoriatic arthritis (PsA) and ulcerative colitis (UC). While JAK inhibition via baricitinib (BARI; JAK-1/2), upadacitinib (UPA; JAK-1/2) and filgotinib (FILGO; JAK-1) also showed good efficacy in different indications, questions on how JAK selectivity influences clinical efficacy as well as the safety profile of all these agents arose and are still insufficiently answered, continue to be issues for debate and future research.15
To guide the practicing clinician on how (not when) to use JAKi in clinical practice, the consensus meeting on ‘Points to consider for the treatment of immune-mediated inflammatory diseases with Janus kinase inhibitors’ was conducted in 2019. Participants of the consensus task force were informed by this systematic literature research (SLR) on the efficacy and safety of all trials on JAKi conducted in IMIDs.
METHODS
A review protocol for this SLR was developed by the steering group in accordance with the EULAR standardised operating procedures for recommendations.16
The systematic literature search was conducted by a database expert (LF) in EMBASE, Medline and the Cochrane Library. The search included all studies published from the earliest date indexed until 12 March 2019 (last date searched). Further, the conference abstract archives of the EULAR Annual Meeting and American college of rheumatology (ACR, until 2018) were hand-searched. All search terms used are shown in the online supplemental appendix (Section 1.1.1–1.1.3). Data extraction was done by one researcher (AK) in duplicates.
The eligibility criteria for inclusion were defined as studies in adult patients (≥18 years) treated with JAKi with diagnosed active autoimmune disease, that is, RA, PsA, AS, systemic lupus erythematosus (SLE), Crohn’s disease (CD), UC, psoriasis (PsO), atopic dermatitis (AD) or alopecia areata (AA) and alopecia universalis. Patient populations were defined for each disease separately, based on treatment history such as an insufficient response (IR) to certain previous systemic therapies. Data of full articles could be included also if published after the last date of the database search, provided at least one abstract of the respective trial had been published within the SLR’s time frame.
For efficacy evaluation, only randomised, controlled, double-blind trials on systemic or topical JAKi including BARI, decernotinib (DEC), FILGO, peficitinib (PEF), RUXO, TOFA and UPA treatment were considered. A detailed list of patient populations, interventions, controls and outcomes is shown in the online supplemental appendix (section 1.4.1.1–1.4.1.4). For safety evaluation, RCTs were evaluated for signals of adverse events (AEs). In addition, cross-sectional, cohort and case–control studies were eligible.
Research questions developed by the steering group are shown in sections 1.4.2 (for efficacy) and 1.4.3 (for safety) in the online supplemental appendix.
Safety outcomes of interest were infections, malignancies, venous thromboembolic events (VTE), haematological abnormalities (anaemia, leucopenia, neutropenia, lymphopenia), MACE and laboratory abnormalities (hepatic, cholesterol, creatine kinase).
As decided by the steering group, due to expected heterogeneity of the populations investigated, no pooling of efficacy or safety results by meta-analysis was conducted.
Risk of bias (RoB) was assessed using the Cochrane Collaboration’s RoB tool for RCTs, assigning each study as having low, unclear or high RoB.17
Supplemental material
RESULTS
A total of 3454 studies were assessed in the title and abstract screening with 262 selected for full article review; 85 publications were finally evaluated in detail. Figure 1 shows the study flow chart with a detailed description of the selection process. Reports on efficacy selected for inclusion are shown in table 1 (detailed results of included articles are shown in online supplemental appendix tables S2.1.1–2.1.9).
Efficacy of Janus kinase inhibitors investigated in randomised controlled trials published until March 2019

PRISMA flow chart for studies on JAKi efficacy and/or safety in inflammatory immune disease, published until March 2019. JAKi, Janus kinase inhibitors; PRISMA, Preferred Reporting Items for Systematic Reviews and Meta-Analyses.
Most of the articles showed a low overall RoB, with few articles considered to be of unclear risk due to insufficient reporting on random sequence generation and allocation procedures. One study was considered to have a high RoB due to dosage unblinding of participants and investigators.18 Details are shown in the online supplemental appendix (tables S2.2.1–S2.2.9).
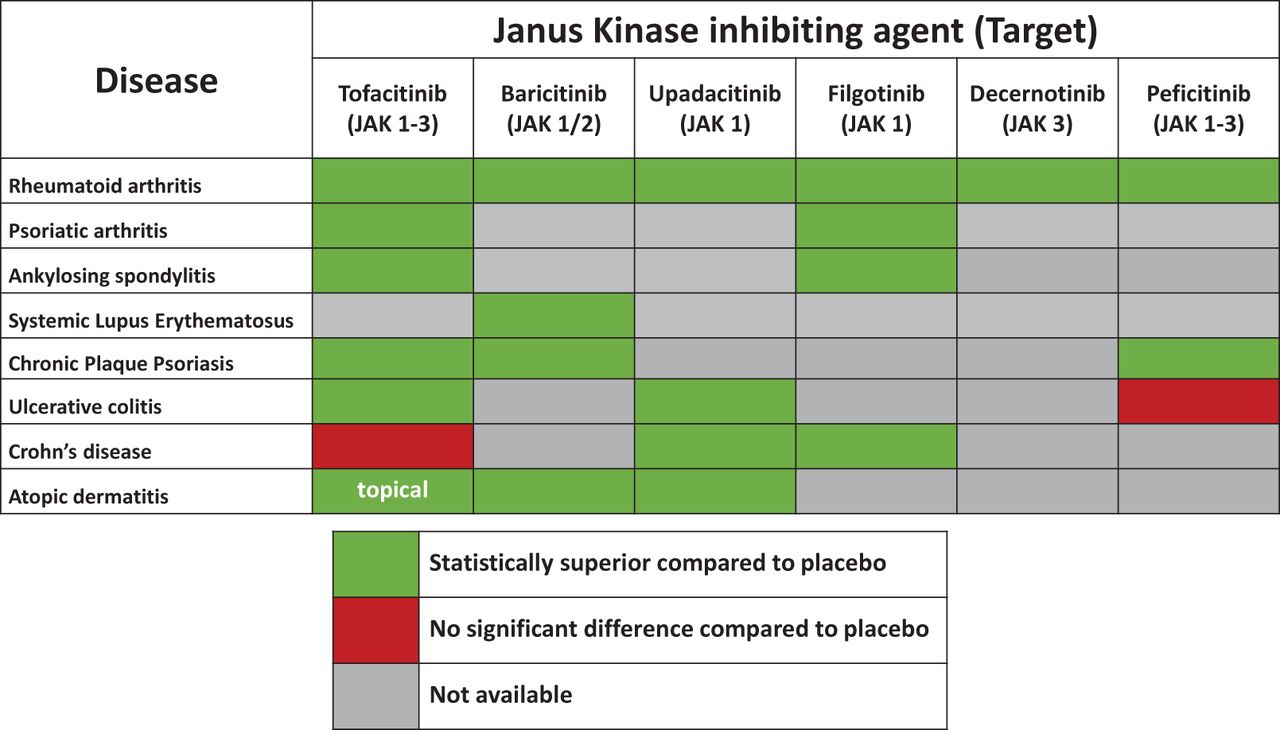
Figure 2 visualises efficacy results of different JAKi by disease, based on the achievement of primary clinical end points. Baseline characteristics (tables S2.3.1–S2.3.9) and detailed efficacy results (tables S3.1–S3.9) are shown in the online supplemental appendix.

{kind=link}
{kind=link}
Efficacy of Janus kinase inhibiting agents across immune-mediated diseases (based on available data at end of March 2019). JAK, Janus kinase.
Besides safety data of clinical trials investigated for efficacy, 13 additional reports on safety were included (details of selected articles are shown in online supplemental appendix tables S4.1.1–S4.1.8; safety outcomes are shown in online supplemental appendix tables S4.2.1–S4.2.8 and S4.3.1–S4.3.8).
Rheumatoid arthritis
In total, 39 primary reports of clinical trials on JAKi in patients with RA were included (low RoB: n=28; unclear RoB: n=9; conference abstracts: n=3; for details on study characteristics, RoB analyses, baseline characteristics and efficacy outcomes, see online supplemental appendix tables S2.1.1, S2.2.1, S2.3.1 and S3.1).
TOFA was effective in reducing signs and symptoms of RA as well as inhibition of radiographic damage progression. These studies were performed in methotrexate (MTX)-naïve patients,19 patients with IR to MTX,20 21 or conventional synthetic disease-modifying drugs (csDMARDs)1 22–27 or to tumour necrosis factor alpha inhibitors (TNFi) and other biological (b)DMARDs1 28 29; structural outcomes were only studied in MTX-naïve and MTX-IR patients. In MTX-IR patients, van Vollenhoven et al showed numerically similar response rates of TOFA and adalimumab (ADA), which was used as an active comparator but not powered for non-inferiority in the ORAL Standard trial.30 ORAL-Strategy, a head-to-head trial comparing TOFA 5 mg two times per day monotherapy and TOFA 5 mg two times per day plus MTX with ADA 40 mg every other week (EOW) plus MTX, proved non-inferiority between the combination therapy arm but not for the TOFA monotherapy arm compared with the two combination arms (table 2).31
Trials investigating Janus kinase inhibitors and tumour necrosis factor alpha inhibitors
Studies on BARI also revealed the efficacy of BARI 2 mg and 4 mg once daily (OD) in csDMARD-naïve,36 MTX and csDMARD-IR,37–40 and patients previously not responding to bDMARDs, compared with placebo.2 In RA-BEAM, BARI 4 mg was statistically superior clinically over placebo and ADA 40 mg EOW in MTX-IR patients (table 2).32 In a randomised tapering substudy of RA-BEYOND, Takeuchi et al showed that patients on BARI 4 mg OD who had achieved low disease activity according to the Clinical Disease Activity Index (CDAI, ≤10) and were subsequently randomised to reduce the BARI dose from 4 mg to 2 mg OD mostly maintained their disease state, although less so than continuing full dose (CDAI≤10 at week 48: continued BARI 4 mg vs tapering to BARI 2 mg: 80% vs 67%; patients achieving CDAI≤2.8, that is, remission: BARI 4 mg vs 2 mg: 40% vs 33%); there was a numerically lower rate of non-serious infections in the tapering arm.41 42
UPA was investigated in eight trials, also provided good efficacy results across various patient populations with RA (MTX-naïve, MTX-IR, csDMARD-IR, TNF-IR and bDMARD-IR) compared to placebo and both as monotherapy and when combined with MTX.3 37 43–50 A head-to-head comparison of UPA+MTX with ADA 40 mg EOW + MTX demonstrated superiority (clinically and functionally) of UPA+MTX versus ADA+MTX and versuss placebo + MTX (table 2).33 34 In SELECT-MONOTHERAPY, MTX-IR patients were either randomised to blinded UPA 15 mg OD, UPA 30 mg OD or continued MTX for 14 weeks. UPA showed statistically superior responses in clinical and functional outcomes, compared to continued MTX (ACR20 at week 14: 68%, 71% and 41% for UPA 15 mg OD, 30 mg OD and continued MTX, respectively).44 45
Treatment with FILGO in MTX-IR patients showed superiority compared to placebo in four phase II RCTs.4 51 52 FILGO monotherapy (DARWIN 2) was superior to placebo after MTX washout (ACR20 at week 12: 67% vs 66% vs 73% vs 29% for FILGO 50 mg OD, 100 mg OD, 200 mg OD and placebo, respectively).4 51 52 FILGO in combination with MTX also showed superiority over placebo + MTX in DARWIN 1 (ACR20 at week 12: 56% vs 64% vs 69% vs 57% vs 60% vs 79% vs 44% for FILGO 50 mg OD, 100 mg OD, 200 mg OD, 25 mg two times per day, 50 mg two times per day, 100 mg two times per day and placebo, respectively).4 DEC (JAK-3 selective) showed superiority over placebo in three trials; however, no clear dose–response relationship exists in ACR responses.53–55 PEF, another pan-JAKi (JAK 1–3), was investigated in two global trials, where it failed to reveal significant efficacy (Genovese 2017: ACR20 at week 12: 22.0% vs 36.8% vs 56.3% vs 29.3%; Kivitz 2017: 43.9% vs 61.5% vs 46.4% vs 57.7% vs 44.4% for PEF 25 mg OD, 50 mg OD, 100 mg OD, 150 mg OD and placebo, respectively). But in several Japanese RA study populations (MTX-naïve, MTX-IR, csDMARD-IR), it showed significant improvement of signs and symptoms and physical function compared to placebo.56–62
PsA, ankylosing spondylitis and SLE
In PsA, three trials (all with low RoB) were published (for details, see online supplemental appendix tables S2.1.2, S2.2.2, S2.3.2 and S3.2). TOFA was investigated in two phase III trials, showing efficacy not only regarding signs and symptoms of arthritis but also physical function, skin disease, dactylitis and enthesitis.5 63 Similar results across many outcomes (although not formally tested) in patients with csDMARD-IR PsA were observed with TOFA compared to ADA 40 mg EOW (table 2).5 Treatment with FILGO 200 mg OD in EQUATOR resulted in significant improvements compared to placebo regarding signs and symptoms of arthritis, PsO and enthesitis.64
In ankylosing spondylitis, two trials in patients with IR to non-steroidal anti-inflammatory drugs were available for analysis, one on TOFA and one on FILGO (for details, see online supplemental appendix tables S2.1.3, S2.2.3, S2.3.3 and S3.3).6 65 In a phase II trial (unclear RoB), TOFA significantly improved clinical outcomes of spinal mobility, pain and function as well as inflammatory changes by MRI, with a clear dose–response.6 In phase II trial (low RoB), FILGO improved disease activity significantly more than placebo across various outcome measures.65
Only limited data on JAKi in SLE were published (two trials, one with high RoB, one with low RoB; for details, see online supplemental appendix tables S2.1.4, S2.2.4, S2.3.4 and S3.4).7 18 In a phase II study, Wallace et al investigated BARI in patients with SLE and active skin or joint disease. Significantly more patients achieved a SLEDAI-2 K resolution of arthritis or rash at week 24 with BARI 4 mg OD (but not BARI 2 mg OD) compared to placebo treatment.7
Chronic plaque PsO, AD and alopecia
Ten trials on patients suffering from chronic plaque PsO were included in this SLR (for details, see online supplemental appendix tables S2.1.5, S2.2.5, S2.3.5 and S3.5), eight with low, one with unclear and one with high RoB, respectively. TOFA showed significant improvements in skin disease compared to placebo in patients who were candidates for systemic therapy or phototherapy.8 35 66 67 Bachelez et al could demonstrate non-inferiority of TOFA 10 mg two times per day compared with etanercept 50 mg twice weekly in achieving a Psoriasis Area and Severity Index (PASI) 75% response as well as clear or almost clear skin (as evaluated by the physician global assessment, PGA) at week 12 (table 2).35 In a withdrawal and retreatment trial, patients with treatment response to TOFA 5 mg or 10 mg two times per day at week 24 were re-randomised to placebo or their previous TOFA dose. Moreover, 23.3% and 26.1% of the patients withdrawn from TOFA 5 mg and 10 mg two times per day, respectively, could maintain their PASI75% response (compared to 56.2% and 62.3% with ongoing TOFA 5 mg or 10 mg two times per day) after 16 weeks. Following 16 weeks of retreatment, 36.8% and 61% of the patients who relapsed after treatment withdrawal, could again achieve a PASI 75% response (compared with 63% and 73.8% of the patients continuously treated with TOFA 5 mg or 10 mg two times per day, respectively).68 A dose-finding study investigating BARI showed BARI 8 mg OD as well as 10 mg OD (but neither 2 mg OD nor 4 mg OD) to be significantly better than placebo in achieving the primary end point (PASI75% at week 12).69 A JAK-1 selective JAKi, itacinib (INCB039110), showed promising results in a 28-day proof-of-concept PsO trial.70 BMS-986165, considered as selective TYK2 inhibitor, showed better clearing of PsO than placebo at week 12.71
Topical TOFA showed greater improvements in pruritus and eczema area and severity compared to vehicle treatment in AD.72 Another topical pan-JAKi (JTE-052) showed rapid and significant AD improvements over vehicle treatment, with numerically similar results to open-label topical tacrolimus, but potential unblinding during the study (high RoB).73
Further, systemic treatment with BARI showed promising results in improving signs, symptoms and patient-reported outcomes of AD.74 Dose-dependent responses to UPA with significant differences compared to placebo were demonstrated in a phase II study in patients with moderate to severe AD (% change from baseline in Eczema Area and Severity Index at week 16: 39% vs 62% vs 74% vs 23% for UPA 7.5 mg OD, 15 mg OD, 30 mg OD and placebo, respectively).9 10
Selective inhibition of JAK-3 via PF‐06651600 and TYK2/JAK1 via PF‐06700841 showed statistically superior results compared to placebo in patients with AA regarding ≥50% improvement from baseline in severity of alopecia tool at 24 weeks (conference abstract, no RoB analysis conducted).75
Inflammatory bowel disease
In total, eleven reports on eight trials in inflammatory bowel disease were included, describing four trials on UC (all with low RoB) and four on CD (3 low RoB, 1 unclear RoB; for details, see online supplemental appendix tables S2.1.7, S2.1.8, S2.2.7, S2.2.8, S2.3.7, S2.3.8, S3.7 and S3.8).11–14 76–82
TOFA 10 mg two times per day was effective in UC as induction therapy compared to placebo in UC, with 16.6% vs 8.2% achieving remission (defined as total Mayo score ≤2, with no subscore >1 and a rectal bleeding subscore of 0) at week 8. Patients with clinical response were subsequently randomised to receive TOFA 10 mg two times per day, TOFA 5 mg two times per day or placebo with TOFA being significantly more effective than placebo therapy after 52 weeks (34.3% vs 40.6% vs 11.1% for TOFA 5 mg two times per day, TOFA 10 mg two times per day and placebo, respectively).76 Induction therapy in patients with moderate to severe UC with UPA was investigated in a phase II trial, being more effective than placebo in inducing remission (0% vs 8.5% vs 14.3% vs 13.5% vs 19.6% for placebo, UPA 7.5 mg, 15 mg 30 mg or 45 mg OD) at week 8.77 78 In a phase IIb study on PEF as induction therapy in UC, the primary end point, establishment of a dose–response relationship was not met. Only PEF 150 mg showed a significant difference (nominal p<0.05) in inducing remission at week 8 compared to placebo (7% vs 15.9% vs 15.9% vs 27.3% vs 15.9% for placebo, 25 mg OD, 75 mg OD, 150 mg OD or 75 mg two times per day).79
TOFA was not effective in treating patients with active CD and insufficient response to glucocorticoids and immunomodulatory agents (including TNFi), neither for induction nor maintenance therapy in three phase II trials.80 81 A phase II dose-finding study on UPA showed clinical as well as endoscopic improvement in moderate-to-severe CD (clinical/endoscopic remission at week 16: 11%/0% vs 13%/10% vs 27%/8% vs 11%/8% vs vs 14%/14% vs 22%/22% for placebo, 3 mg , 6 mg , 12 mg two times per day, 24 mg OD and 24 mg two times per day, respectively).12 14 After 16 weeks of induction therapy, patients were re-randomised to either UPA 3 mg, 12 mg or 24 mg two times per day for maintenance therapy up until week 52. A dose–response in clinical as well as endoscopic outcomes was shown over 36 weeks of treatment in initial responders of the induction phase (clinical/endoscopic remission at week 52: 41.2%/25% vs 62.5%/25% vs 73.3%/37.5% vs 40%/10% for UPA 3 mg , 6 mg , 12 mg two times per day and 24 OD).13 14 JAK-1 selective treatment with FILGO 200 mg OD resulted in significantly more patients achieving clinical remission (Crohn’s Disease Activity Index <150 at week 10: 47% vs 23% for FILGO 200 mg OD vs placebo). Results on endoscopic outcomes were numerically higher, although not statistically different at week 10 (50% response: 25% vs 14%, remission: 14% vs 7% at week 10, for FILGO 200 mg OD and placebo, respectively).82 Detailed results of trials investigating JAKi and TNFi are shown in table 2.
Safety
All RCTs with a valid comparator (placebo or active treatment arm in the respective time period) were assessed for AEs of special interest (see online supplemental appendix tables S4.1.1–S4.1.8 for details of included reports). Numerically higher rates of serious AEs, infections, serious infections and especially herpes zoster (HZ) were identified when comparing JAKi treatment arms with placebo arms (see online supplemental appendix tables S4.2.1–S4.2.8 for detailed results). AEss of special interest are shown in online supplemental appendix tables S4.3.1–S4.3.8. Liver enzymes and creatine kinase elevations as well as grade 3 or 4 lymphopenia/leucopenia appeared more frequently during JAKi treatment.
Assessment of rare events, especially VTE (that is, deep vein thrombosis and pulmonary embolism) remained difficult to assess due to scarcity of data and a largely diverse database on the amplitude of VTE risk related to the underlying disease. One cohort study using claims databases investigated the risk of venous thromboembolism in biological and JAKi-naïve patients with RA (n=50 865) receiving TOFA versus patients receiving TNFi and found a numerically higher, however, statistically nonsignificant VTE risk (pooled propensity score-adjusted HR: 1.33; 95% CI 0.78 to 2.24).83
In some RCTs, compared to placebo, numerically higher rates of DVT/PE were seen in JAKi-treated patients, suggesting an increased risk for venous thromboembolism. However, reports of head-to-head studies did not reveal a clear signal regarding VTE risk when comparing TNFi and JAKi treatment arms during the controlled period (table 3). While the individual reports did not allow to discern major differences between JAKi and placebo or active treatment arms regarding VTEs and PEs, the regulators have published important information after the time point of this SLR. For completeness, we refer to the Food and Drug Adminstration report revealing more VTE/PE for BARI at 4 mg,84 and to the EMA report on a still ongoing trial of TOFA versus anti-TNFs in patients with RA with cardiovascular risk factors where significantly more VTE/PE and deaths were seen with TOFA 10 mg two times per day but numerically also for TOFA 5 mg.85 86 These important information also led to black box warnings in the package inserts of marketed JAKi and more long-term data will be needed, as also meanwhile partly published for some of the drugs.87–90
Adverse events of special interest in randomised controlled trials investigating Janus kinase inhibitors and tumour necrosis factor alpha inhibitors
No safety signal could be identified regarding major cardiac adverse events (MACE), malignancies excluding non-melanoma skin cancer. Table 3 shows AEs of special interest in trials comparing JAKi next to TNFi and placebo treatment.
Additionally, 12 reports on integrated safety analyses of RCTs+long-term extension trials (LTEs) were included in the analysis. TOFA RCTs (until March 2015) comprising 19 406 patient-years (PY) of treatment exposure within RA showed stable AEs over time (median exposure 3.4 years). Nasopharyngitis, upper respiratory tract infections and urinary tract infections were the most common AEs. Most common serious infections were pneumonia, HZ, urinary tract infections and cellulitis, with baseline glucocorticoid usage, age and geographic region (Asia) being significant risk factors. No increased incidence rate for malignancies excluding non-melanoma skin cancer (standardised incidence ratio: 1.0; 95% CI 0.8 to 1.1) was observed. Twenty-two gastrointestinal perforations (incidence ratio: 0.11, 95% CI 0.07 to 0.17) were reported, all in patients with concomitant non-steroidal anti-inflammatory drugs (NSAIDs) or glucocorticoid therapy (NSAIDs + glucocorticoids: n=10; NSAIDs only: n=9; glucocorticoids only: n=3); 13 patients had a history of diverticulitis or diverticulosis and two a history of gastric ulcers.91 Consistent results were also observed in patients with UC and PsO who were treated with TOFA in RCTs and LTEs.92 93 Safety analyses on HZ events in patients with PsO and UC found patients treated with TOFA at increased risk for HZ infection, with age, Asian origin and previous biological use as risk factor as well as dose-dependent higher risks in patients treated with TOFA 10 mg two times per day versus TOFA 5 mg two times per day.94–96 Although higher levels of low-density lipoprotein, high-density lipoprotein and total cholesterol were observed during TOFA treatment, no signal regarding a higher risk of MACE was found in RA, PsO and UC trials.97–99
Integrated safety on BARI in RA with 6637 total PY of exposure (median 2.1 years) showed a higher risk for infections including HZ; VTE (including deep vein thrombosis/pulmonary embolism) were reported with BARI 4 mg OD but not for placebo (IR 0.5/100PY, 95% CI 0.3 to 0.7) without differences between BARI 2 mg (IR 0.5/100PY) and BARI 4 mg (IR 0.6/100 PY). These were associated with age, higher BMI, history of DVT/PE and use of selective cyclooxygenase-2 inhibitors. Higher rates of non-melanoma skin cancer were identified in BARI 4 mg OD compared to BARI 2 mg OD-treated patients. Three cases of gastrointestinal perforations were reported in patients taking MTX+NSAIDs, with two patients taking glucocorticoids. Ten cases of tuberculosis (in endemic areas) were observed in BARI-treated patients. No increased risk of MACE or malignancies was identified.100
Pregnancy is a contraindication for JAKi therapy, and patients were required to use contraception during the RCTs. Therefore, only very limited data (two retrospective analyses) on pregnancy outcomes were available.101 102 Clowse et al investigated pregnancy outcomes of patients treated with TOFA in RA (31 maternal cases: TOFA monotherapy n=18, TOFA+MTX n=13; 3 paternal cases) and PsO (16 maternal cases, 41 paternal cases). Similar frequencies of healthy newborns (n=25), no fetal death, seven spontaneous abortions, eight medical terminations and one congenital malformation (pulmonary valve stenosis) were reported until April 2014. These frequencies were consistent with background risks in the general population as well as in patients with RA or PsO, although confounded through concomitant MTX therapy in some patients with RA.101 Further analysis on pregnancies in UC, RA, PsO and PsA RCTs (up to March 2017) reported results in line with the previous report with pregnancy AEs during TOFA treatment appearing similar to those in the general population.102
DISCUSSION
We conducted this SLR to inform the task force on points to consider for the treatment of IMIDs with JAKi with data of all reports and conference abstracts published until March 2019.
Efficacy of JAK inhibition has been shown for several agents being either pan-JAKi (TOFA, PEF) as well as JAK-selective (BARI, UPA, FILGO, DEC) compounds. With TOFA being the first and therefore most extensively studied agent, the JAKi approved to date demonstrating good efficacy in various indications. FILGO recently received a positive opinion by the European Medicines Agency, recommending the granting of a marketing authorisation for treatment of RA. However, none of the available JAKi was approved for PsO until now, as efficacy data were especially promising in higher doses, but these were not approved due to regulatory safety concerns. The JAKi approved up until the date of submission of this manuscript (TOFA, BARI, UPA) appeared to demonstrate a similar safety profile with an increased risk of infections (particularly HZ) and a potential risk of VTE. Although overall rare, VTE were observed in patients at risk for thrombosis, subsequently leading to warnings issued by the regulators. However, large registry data and studies of at-risk patients with sufficiently large cohort and comparator arms for safety analyses are still lacking.
There are several limitations of this SLR: (1) only one researcher (AK) conducted the title and abstract screening, data extraction and RoB analysis; (2) we only reported data narratively due to the heterogeneity of data; (3) safety analyses were completely based on RCTs and their LTEs, limiting the interpretability due to selection bias of clinical trial patient populations, which are only partly comparable to the general population; (4) RoB is difficult to assess in conference abstracts (and was therefore not assessed). Possibly, data of conference abstracts of trials with poor study design and/or inconsistent or incomplete results may therefore never get published in peer-reviewed journals. However, in advance of the meeting, critical questions regarding the SLR could be discussed with members of the task force with long-standing experience in clinical trials of JAKi, including efficacy and safety analyses, and re-checked in the literature when needed.
This SLR formed the basis for the formulation of the points to consider for the treatment of IMIDs with JAKi and for the definition of the levels of evidence and strengths of recommendations of each item.103
Key messages
What is already known about this subject?
Numerous randomised controlled trials investigating the efficacy and safety of Janus kinase inhibitors in immune-mediated inflammatory diseases have been conducted.
What does this study add?
JAKi were effective in reducing signs and symptoms in rheumatic diseases (rheumatoid and psoriatic arthritis, ankylosing spondylitis and systemic lupus erythematosus) as well as inflammatory bowel disease (ulcerative colitis and Crohn’s disease) and immune-mediated dermatological diseases like chronic plaque psoriasis and atopic dermatitis.
Janus kinase inhibitors showed an acceptable safety profile in the investigated populations, with an increased risk for infections (including serious infections and herpes zoster). Rare events were difficult to assess for some agents due to the limited amount of patient-exposure-years and only few registry data. However, numerically higher rates of venous thromboembolic events were seen in JAKi-treated patients in some studies.
How might this impact on clinical practice?
This SLR was performed to inform the task force on ‘Points to consider for the treatment of immune-mediated inflammatory diseases with Janus kinase inhibitors’ with the evidence published until March 2019.
REFERENCES
Footnotes
Contributors All authors contributed and finally approved the current manuscript.
Funding This study was supported by grants from AbbVie and Lilly. The companies had no influence on the selection of participants, were not present at any of the meetings and had no influence on the contents of the present paper.
Competing interests AK: Speakers bureau: Bristol-Myers Squibb, Celgene, Eli Lilly, Merck Sharp and Dohme, Novartis, Pfizer; non-financial support: Gilead. JSS: Amgen, AbbVie, AstraZeneca, Astro, BMS, Celgene, Glaxo, ILTOO, Janssen, Merck-Serono, MSD, Novartis-Sandoz, Pfizer, Roche-Chugai, Samsung, UCB. PN: AbbVie, BMS, UCB, Lilly, Gilead/Galapagos, Pfizer, GSK, Roche, Sanofi, Janssen, MSD, Novartis, Boehringer-Ingelheim, Celgene, Samsung. TD: AbbVie, BMS, Celgene, Eli Lilly, Janssen, EMD Merck-Serono, Galapagos, Gilead, Novartis, Roche, Samsung, UCB. MD: AbbVie, Biogen, Celgene, Janssen, Lilly, Novartis, Merck, Pfizer, Sanofi-Aventis, UCB. RF: Consultant: AbbVie, Acea, Akros, Amgen BMS, Celltrion, Gilead, GSK, Jansen, Eli Lilly, Novartis, Pfizer, Samsung, Sanofi-Aventis, Tahio, UCB; Data Safety Monitoring Boards EMDSerano, Celltrion; Clinical Trial Grants: AbbVie, Acea, Akros, Amgen, AstraZeneca, BMS, Gilead, GSK, Janssen, Eli Lilly, Novartis, Pfizer, Regeneron, Sanofi-Aventis, UCB. KG has received consultancy and lecture fees from Novartis, Pfizer and Roche and investigational grants from Roche. IBM has received research funding or honoraria from AbbVie, AstraZeneca, Celgene, GSK, Lilly, Boehringer, Pfizer, Janssen, Novartis, UCB, BMS, Sanofi. TT: AbbVie GK, Astellas, Asahi Kasei, Chugai, Daiichi Sankyo, Eisai, Mitsubishi Tanabe, Pfizer Japan, Nippon Kayaku and Takeda. MT: speaker fees from Bristol-Myers Squibb (BMS), Falk Foundation, Gilead, Intercept and Merck Sharp & Dohme (MSD); advisory board fees from Albireo, Boehringer Ingelheim, BiomX, Falk Pharma GmbH, GENFIT, Gilead, Intercept, MSD, Novartis, Phenex, and Regulus; travel grants from AbbVie, Falk, Gilead, and Intercept; and research grants from Albireo, CymaBay, Falk, Gilead, Intercept, MSD, and Takeda. He is also coinventor of patents on the medical use of norUDCA filed by the Medical University of Graz. KW: Research grants from BMS, Pfizer; Consulting fees: AbbVie, BMS, Eli Lilly, Gapalagos, Gilead, Pfizer, UCB, Regeneron, GSK, and Roche; MdW has received honoraria for consultancies and speaking through Stichting Tools from AbbVie, BMS, Celgene, Janssen, Lilly, Novartis, Pfizer, Roche. W-HB has received a research grant from Pfizer and honoraria for advice from AbbVie, Alimirall, BMS, Celgene, Janssen, Leo, Lilly, Novartis and UCB. LF: Nothing to declare. DvdH: Consulting fees: AbbVie, Amgen, Astellas, AstraZeneca, BMS, Boehringer Ingelheim, Celgene, Cyxone, Daiichi, Eisai, Eli Lilly, Galapagos, Gilead, GSK, Janssen, Merck, Novartis, Pfizer, Regeneron, Roche, Sanofi, Takeda, UCB Pharma, Director of Imaging Rheumatology BV.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplemental information.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.