Article Text

Abstract
Objective Tofacitinib is an oral Janus kinase (JAK) inhibitor for the treatment of rheumatoid arthritis (RA). We report the largest integrated safety analysis of tofacitinib, as of March 2017, using data from phase I, II, III, IIIb/IV and long-term extension studies in adult patients with RA.
Methods Data were pooled for patients with RA who received ≥1 tofacitinib dose. Incidence rates (IRs; patients with events/100 patient-years [PY]; 95% CIs) of first-time occurrences were obtained for adverse events (AEs) of interest.
Results 7061 patients received tofacitinib (total exposure: 22 875 PY; median [range] exposure: 3.1 [0 to 9.6] years). IRs (95% CI) for serious AEs, serious infections, herpes zoster (all), opportunistic infections (excluding tuberculosis [TB]) and TB were 9.0 (8.6 to 9.4), 2.5 (2.3 to 2.7), 3.6 (3.4 to 3.9), 0.4 (0.3 to 0.5) and 0.2 (0.1 to 0.2), respectively. IRs (95% CI) for malignancies (excluding non-melanoma skin cancer [NMSC]), NMSC and lymphomas were 0.8 (0.7 to 0.9), 0.6 (0.5 to 0.7) and 0.1 (0.0 to 0.1), respectively. IRs (95% CI) for gastrointestinal perforations, deep vein thrombosis, pulmonary embolism, venous thromboembolism, arterial thromboembolism and major adverse cardiovascular events were 0.1 (0.1 to 0.2), 0.2 (0.1 to 0.2), 0.1 (0.1 to 0.2), 0.3 (0.2 to 0.3), 0.4 (0.3 to 0.5) and 0.4 (0.3 to 0.5), respectively. IR (95% CI) for mortality was 0.3 (0.2 to 0.3). IRs generally remained consistent across 6-month intervals to >78 months.
Conclusion This represents the largest clinical dataset for a JAK inhibitor in RA to date. IRs remained consistent with previous reports from the tofacitinib RA clinical development programme, and stable over time.
Trial registration numbers NCT01262118; NCT01484561; NCT00147498; NCT00413660; NCT00550446; NCT00603512; NCT00687193; NCT01164579; NCT00976599; NCT01059864; NCT01359150; NCT02147587; NCT00960440; NCT00847613; NCT00814307; NCT00856544; NCT00853385; NCT01039688; NCT02187055; NCT00413699; NCT00661661.
For summary of phase I, phase II, phase III, phase IIIb/IV and LTE studies included in the integrated safety analysis, see online supplemental table 1.
- Arthritis
- Rheumatoid
- Therapeutics
- Antirheumatic Agents
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
The efficacy and safety of tofacitinib 5 and 10 mg twice daily have been demonstrated in patients with moderately to severely active rheumatoid arthritis (RA) in previous phase II, III and IIIb/IV randomised controlled trials (RCTs) and open-label, long-term extension (LTE) studies. Safety was generally similar with tofacitinib, compared with biological disease-modifying antirheumatic drugs (bDMARDs); however, an increased risk of certain types of infection (eg, herpes zoster [HZ]) was observed.
An integrated analysis of the long-term safety of tofacitinib in patients with RA was carried out in 2015, using RCT/LTE data, with up to 8.5 years of tofacitinib exposure. Adverse events (AEs) were generally stable over time; no new safety risks were observed, compared with previous analyses of tofacitinib safety.
What does this study add?
This integrated safety summary of completed RCTs and LTE studies, using data up to 2017, spans 9.5 years of cumulative tofacitinib exposure in >7000 patients, and represents the largest clinical dataset for a Janus kinase (JAK) inhibitor in RA to date.
Overall, incidence rates (IRs; patients with events/100 patient-years [PY] of exposure) of AEs of special interest, discontinuations due to AEs and mortality with tofacitinib were consistent with those reported previously. With the exception of HZ (non-serious and serious), serious infections, malignancies (excluding non-melanoma skin cancer [NMSC]) and NMSC, IRs for AEs of special interest (including thromboembolic events) were <0.5/100 PY.
How might this impact on clinical practice?
This analysis is the largest integrated safety analysis of tofacitinib to date. With the exception of HZ, rates of safety events were generally similar with tofacitinib, compared with bDMARDs and other JAK inhibitors used to treat RA.
INTRODUCTION
Rheumatoid arthritis (RA) is a chronic and debilitating autoimmune disease affecting approximately 0.24% of the global population.1 Current treatment guidelines recommend biological disease-modifying antirheumatic drugs (bDMARDs), such as tumour necrosis factor inhibitors (TNFi), or targeted synthetic DMARDs, such as Janus kinase (JAK) inhibitors, in patients who have failed treatment with conventional synthetic DMARDs (csDMARDs).2 3
Supplemental material
Tofacitinib is an oral JAK inhibitor for the treatment of RA. The efficacy and safety of tofacitinib 5 and 10 mg twice daily (BID) administered as monotherapy or in combination with csDMARDs, mainly methotrexate (MTX), in patients with moderately to severely active RA, have been demonstrated in phase II,4–8 phase III9–14 and phase IIIb/IV15 studies of up to 24 months’ duration, and in long-term extension (LTE) studies with up to 114 months’ observation.16–18 Tofacitinib was first approved by the US Food and Drug Administration (FDA) in 2012 for the treatment of patients with RA.19 In February 2019, the Data Safety Monitoring Board for tofacitinib rheumatology studies determined that the frequency of pulmonary embolism (PE) and all-cause mortality in the tofacitinib 10 mg BID arm was higher than in the TNFi comparator arm in an FDA post-marketing requirement safety study (A3921133; NCT02092467; database not locked and data have not yet been source verified or subjected to standard quality check procedures that would occur at database lock and may therefore be subject to change)20 designed to evaluate the long-term risk of major adverse cardiovascular events (MACE) and malignancy. Study A3921133 is an ongoing, open-label, endpoint-driven study, evaluating the safety of tofacitinib 5 and 10 mg BID versus TNFi in patients with RA. To be eligible for enrolment, patients had to be ≥50 years of age, have at least one cardiovascular (CV) risk factor and be on a stable dose of MTX. Subsequently, based on the information from an ad hoc safety analysis of Study A3921133 and consideration of information pertaining to PE for other JAK inhibitors, venous thromboembolic events (deep vein thrombosis [DVT]/PE) are considered an important identified risk for treatment with tofacitinib.
While the safety profile of the JAK inhibitors to date is generally similar to that of bDMARDs, some differences have been reported, including an increased risk of certain types of infection, most notably herpes zoster (HZ),21 and thromboembolic events.22 A comprehensive review of the long-term safety of tofacitinib was previously carried out using a 2015 integrated analysis of data from >6000 adult patients with RA with a cumulative tofacitinib exposure of up to 8.5 years.23 This study reported that adverse events (AEs) were generally stable over time, and no new safety risks were observed compared with those reported in previous phase I, II, III and IIIb/IV randomised controlled trials (RCTs) and open-label LTE studies in the tofacitinib RA clinical development programme.23
Here, we report an updated and comprehensive integrated safety summary of completed RCTs and LTE studies spanning 9.5 years of cumulative tofacitinib exposure in >7000 patients. This 2017 analysis reports the results of the largest, long-term safety database for a JAK inhibitor in RA and captures the maximum level of patient exposure to tofacitinib from the main LTE study, ORAL Sequel (NCT00413699), which was completed in March 2017. This analysis included all patients meeting individual study inclusion/exclusion criteria, irrespective of potential CV risk factors (ie, patients were not required to be ≥50 years of age or have ≥1 CV risk factor) and did not include ongoing Study A3921133.
METHODS
Patients and study design
Details of the methods have been described previously.23
Data, up to March 2017, were pooled from patients with RA who received ≥1 tofacitinib dose, as monotherapy or with background csDMARDs, across the completed 2 phase I, 10 phase II, 6 phase III, 1 phase IIIb/IV index studies and 2 open-label LTE studies (online supplemental table 1). Safety data are reported up to 114 months.
Adult patients (aged ≥18 years) with a diagnosis of active RA, based on the American College of Rheumatology 1987 Revised Criteria24 and active disease at screening and baseline, were included in index studies. Patients who completed an index study were eligible for inclusion in the LTE studies. Inclusion and exclusion criteria for the index and LTE studies have been previously reported.23
Studies were conducted in accordance with the Declaration of Helsinki and the Good Clinical Practice Guidelines, along with applicable local country regulations and laws. The study protocols were approved by the Institutional Review Boards and/or Independent Ethics Committee at each centre. All patients provided written informed consent.
Dosing
Across the index studies, patients received tofacitinib 1, 3, 5, 10, 15 or 30 mg BID or 20 mg once daily, as monotherapy or with background csDMARDs, mainly MTX (online supplemental table 1).
Data for combined tofacitinib 5 and 10 mg BID populations (all tofacitinib doses) and average dosing are reported (full details in online supplemental methods). Tofacitinib and concomitant RA treatment dose adjustments were allowed at the investigator’s discretion.
Data collection, coding and adjudication
Patients were included in the safety analysis if they had received ≥1 tofacitinib dose. Data for all treatment-emergent AEs and serious AEs (SAEs) occurring during treatment and within 28 days after discontinuation of tofacitinib (the clinical trial observation period) were collected and coded using the Medical Dictionary for Regulatory Activities (MedDRA) v.20.0. Details of baseline comorbidities were also collected. Deaths occurring during treatment and within 28 days after discontinuation of tofacitinib are reported.
Adjudication of events has been described previously23 25 and is summarised in the online supplemental methods.
Statistical analyses
All safety analyses were based on observed data. Incidence rates (IRs) and 95% CIs (calculated via the Exact Poisson method adjusted for exposure time) were based on the number of unique patients (per 100 patient-years [PY] of exposure) with first events during the time between the first and last dose plus 28 days, divided by the time accrued during the risk period (ie, between the first and last dose plus 28 days, or the time accrued to the first event, whichever occurred earlier). Events reported after the 28-day post-treatment final visit (study conclusion) were not included in the IR analysis but were presented in the data listings.
To assess changes in IRs over time, rates were examined within 6-month intervals of tofacitinib exposure.
Additional details on the statistical methods used to analyse risk factors are provided in the online supplemental methods.
The large number of participants in the tofacitinib RA clinical development programme accrued a substantial amount of exposure time. Therefore, to enable future comparison to the AE rates from other RA clinical trial programmes (eg, indirect standardisation), highly stratified distributions of exposure time according to age group, geographical region, prior bDMARD exposure and combination therapy versus monotherapy are provided in the online supplemental data.
RESULTS
Patients
This analysis included 7061 patients, representing 22 875 PY of tofacitinib exposure, with a median exposure of 3.1 years. Overall, 4895 (69.3%), 4055 (57.4%), 3543 (50.2%) and 2740 (38.8%) patients received tofacitinib for ≥12, 24, 36 and 48 months, respectively. Patient baseline demographics and disease characteristics were generally similar between all groups (table 1).
Patient baseline demographics and disease characteristics
Adverse events and serious adverse events
The most common treatment-emergent AE by MedDRA system organ class (SOC) was infection and infestations (56.2% [3970/7061]), and the four most common treatment-emergent AEs by preferred term were viral upper respiratory tract infection (17.3% [1221/7061]), upper respiratory tract infection (17.2% [1214/7061]), urinary tract infection (11.8% [832/7061]) and bronchitis (11.3% [800/7061]). A total of 1857 (26.3%) patients experienced SAEs (IR 9.0 [95% CI 8.6 to 9.4]). A total of 1634 (23.1%) patients discontinued due to AEs (IR 7.1 [95% CI 6.8 to 7.5]) (table 2). A total of 59 deaths were reported (0.8%; IR 0.3 [95% CI 0.2 to 0.3]) (table 2). The most common causes of death by MedDRA SOC were cardiac disorders (n=20, 0.28%), infections and infestations (n=18, 0.25%) and respiratory, thoracic and mediastinal disorders (n=16, 0.23%) (full details in the online supplemental results).
IRs (95% CI) of AEs and SAEs (all-cause)
Serious infections
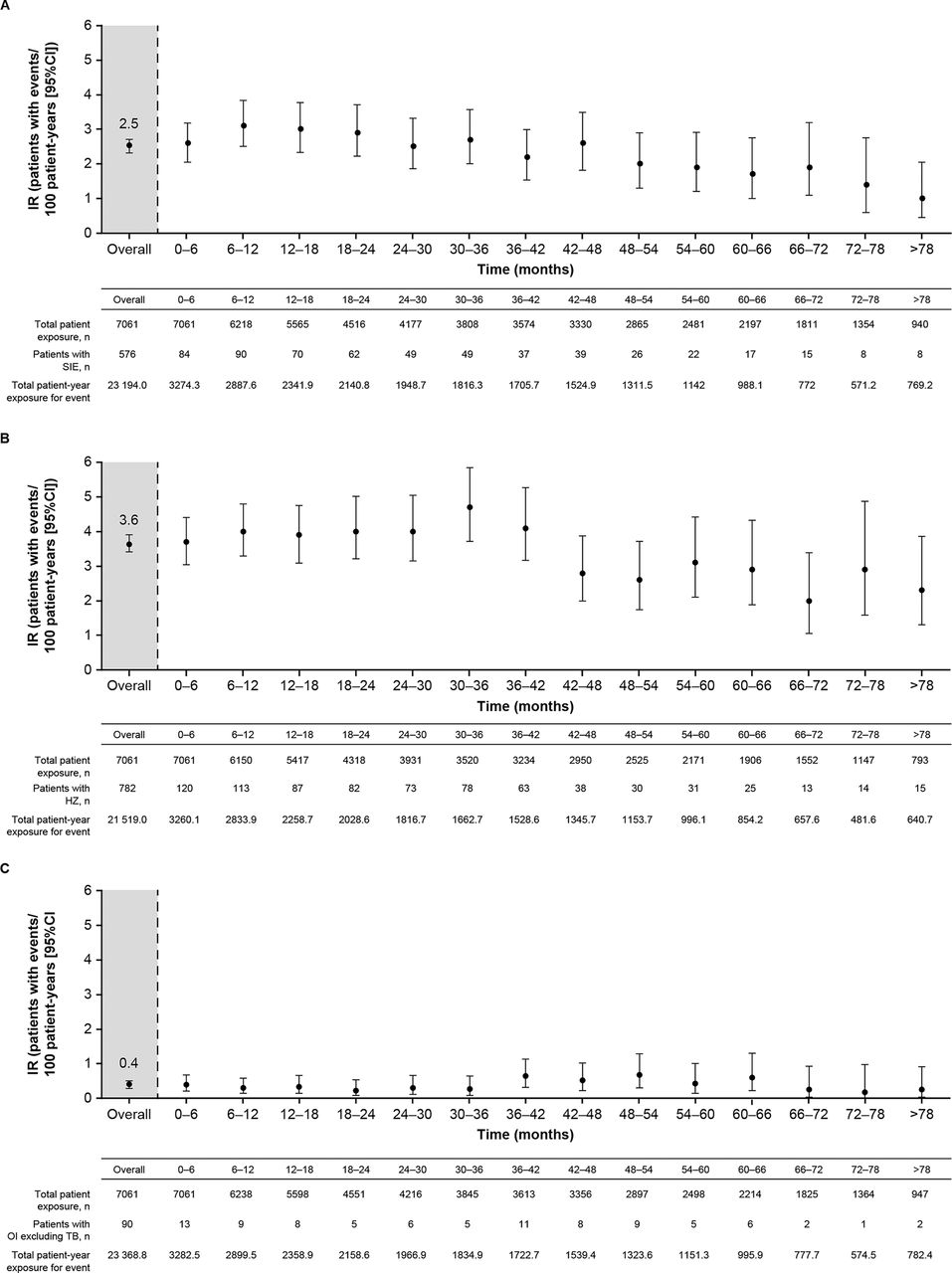
Serious infection events (SIEs) occurred in 576 (8.2%) patients with an IR (95% CI) of 2.5 (2.3 to 2.7). The most common SIEs were pneumonia (n=124), HZ (n=43), urinary tract infection (n=31) and cellulitis (n=31). The IR (95% CI) of SIEs (excluding serious HZ) was 2.3 (2.1 to 2.5). The IRs of SIEs gradually decreased over time with longer exposure to tofacitinib (figure 1A). IRs of SIEs were similar for average tofacitinib 5 and 10 mg BID (table 3). When stratified by prior confirmed lymphocyte counts (two sequential observations), the IRs (95% CI) for SIEs were 7.1 (2.6 to 15.5) for <500 cells/µL, 2.9 (2.5 to 3.5) for ≥500–<1000 cells/µL, 2.4 (2.1 to 2.7) for ≥1000–<1500 cells/µL and 2.3 (2.0 to 2.8) for ≥1500–<2000 cells/µL.
IRs for AEs of interest (95% CI)

IRs for (A) SIEs, (B) HZ (non-serious and serious) and (C) OIs (excluding TB) over time, for all tofacitinib doses (first events). HZ, herpes zoster; IR, incidence rate; OI, opportunistic infection; SIE, serious infection event; TB, tuberculosis.
A Cox regression analysis revealed that factors showing a significantly elevated hazard ratio (HR) for risk of SIEs were tofacitinib dose, higher age, male sex, geographical region (in particular Asia and Australia/New Zealand/rest of world vs USA/Canada), increasing baseline Health Assessment Questionnaire-Disability Index score, prior confirmed post-baseline lymphopenia (defined as prior confirmed cases <1000 cells/µL), baseline glucocorticoid use (all doses vs no use), increasing body mass index, diabetes mellitus and chronic obstructive pulmonary disease (COPD) (all p<0.05) (figure 2A).

HRs of potential risk factors for (A) SIEs (including post-baseline lymphopenia <500 and <1000 cells/µL), (B) HZ (non-serious and serious; including post-baseline lymphopenia <500 cells/µL) and (C) OIs (excluding TB; including post-baseline lymphopenia <500 cells/µL), for all tofacitinib doses. Results from multivariable Cox regression models in the phase I, II, III, IIIb/IV and open-label LTE studies. *This is a time-varying continuous variable (ie, a patient’s dose could be varied during the course of the analysis). †In Unit=x, ‘x’ is the change in the continuous variable corresponding to which the change in hazards is observed. §Medical history and/or complication of COPD. Aus, Australia; COPD, chronic obstructive pulmonary disease; HAQ-DI, Health Assessment Questionnaire-Disability Index; HR, hazard ratio; HZ, herpes zoster; LTE, long-term extension; NZ, New Zealand; OI, opportunistic infection; ROW, rest of world; SIE, serious infection event; TB, tuberculosis.
Herpes zoster
Overall, 782 (11.1%) patients developed HZ, with an IR (95% CI) of 3.6 (3.4 to 3.9). The majority of cases (90.2%) involved a single dermatome. Disseminated HZ was reported in 8 patients (1% of all patients with HZ; all cutaneous, except one case of ocular disease), multi-dermatomal HZ in 43 patients (5.5% of all patients with HZ) and serious HZ in 57 patients (7.3% of all patients with HZ; 0.8% of the total population). Baseline factors with a significant increase in HR for risk of HZ were tofacitinib dose, higher age, geographical region (in particular, Asia vs USA/Canada), smoking (ex-smoker vs never smoked) and baseline glucocorticoid doses (all doses vs no use) (all p<0.05) (figure 2B; online supplemental figure 1A). The crude IRs for HZ were similar for average tofacitinib 5 and 10 mg BID (table 3), but after multivariable adjustment and considering tofacitinib dose as exposed (ie, time-varying), there was a 40% increase in relative risk for HZ associated with an increase of every 5 mg BID dose (figure 2B). Analysis of IRs over 6-month intervals did not show any increase in HZ with longer exposure to tofacitinib (figure 1B). IRs (95% CI) for HZ were higher in Asia than in non-Asian regions (USA/Canada, Europe, Latin America) (5.6 [5.0 to 6.3] vs 3.0 [2.8 to 3.3]), respectively; online supplemental table 2), mainly due to elevated IRs in Japan and the Republic of Korea.
Sixty-two (7.9%) patients had recurrence of HZ (IR 3.7 [95% CI 2.8 to 4.7]), comprising 18 (6.7%; IR 3.0 [95% CI 1.8 to 4.7]) in the tofacitinib 5 mg BID group and 44 (8.6%; IR 4.1 [95% CI 3.0 to 5.5]) in the tofacitinib 10 mg BID group.
Opportunistic infections
Opportunistic infections (OIs; excluding TB) were reported in 90 (1.3%) patients, with an IR (95% CI) of 0.4 (0.3 to 0.5). IRs for average tofacitinib 5 and 10 mg BID were similar and are presented in table 3. IRs for OIs (excluding TB) remained stable with increasing tofacitinib exposure (figure 1C). A list of all OIs (excluding TB) are provided in online supplemental table 3. Most OIs were HZ (57 of 90 cases). There were five cryptococcosis cases: four cryptococcal pneumonia cases and one cryptococcal meningitis case, which resolved.
A Cox regression analysis showed that factors with a significant increase in HR for risk of OIs (excluding TB) were tofacitinib dose, higher age, geographical region (Asia vs USA/Canada), COPD and prior confirmed post-baseline lymphopenia <1000 cells/µL (figure 2C; online supplemental figure 1B).
Tuberculosis
Tuberculosis (TB) is reported separately from other OIs given varying background rates based on geographical region. Active TB was reported in 38 (0.5%) patients, with an IR (95% CI) of 0.2 (0.1 to 0.2). IRs for TB were similar for average tofacitinib 5 and 10 mg BID (table 3). Extrapulmonary/disseminated TB occurred in 19 patients (one of these events was not included in the IR calculation as it occurred outside the 28-day risk period window). The majority of cases (28/38) occurred in geographical regions with a high prevalence of TB (online supplemental table 4). Latent TB was reported in 130 and 190 patients receiving tofacitinib 5 and 10 mg BID, respectively; of these, 121 and 173 patients, respectively, were reported to have been adequately treated with prophylaxis. Four patients who had completed prophylaxis and were randomised to tofacitinib 10 mg BID developed TB during the study period; of these, one patient still had active TB at study completion.
Malignancies
Malignancies (excluding non-melanoma skin cancer [NMSC]) occurred in 177 (2.5%) patients (IR [95% CI] of 0.8 [0.7 to 0.9]) and NMSC in 129 (1.8%) patients (IR 0.6 [95% CI 0.5 to 0.7]); IRs for both were similar regardless of tofacitinib dose (table 3). An analysis of IRs for malignancies (excluding NMSC) and NMSC at 6-month intervals showed that they were generally consistent over time with the overall IRs (figure 3A and B). Outside the 28-day risk period window, where patients were no longer followed in the study and could be receiving alternative therapy, 28 cases of malignancies (excluding NMSC) were reported (not included in the IR calculation) and no cases of NMSC were reported.

{kind=link}
{kind=link}
{kind=link}
IRs for (A) malignancies (excluding NMSC) and (B) NMSC over time, for all tofacitinib doses. IR; incidence rate; NMSC, non-melanoma skin cancer.
The age- and sex-adjusted standardised incidence ratio (SIR) (95% CI) for all malignancies (excluding NMSC) was 0.8 (0.7 to 1.0) using US National Cancer Institute Surveillance and Epidemiology and End Results (SEER) and 1.4 (1.2 to 1.6) using Global Cancer Incidence, Mortality and Prevalence (GLOBOCAN) estimates.
In total, 12 lymphoma events were reported within the 28-day risk period, 1 event in patients receiving an average tofacitinib dose of 5 mg BID (IR 0.01 [95% CI 0.00 to 0.07]) and 11 events occurring in patients receiving an average tofacitinib dose of 10 mg BID (IR 0.07 [95% CI 0.04 to 0.13]) in the main analysis (table 3). Outside the 28-day risk period window used in the calculation of IRs, as a supplemental analysis, a total of 19 lymphoma events occurred, 6 events in patients receiving an average tofacitinib dose of 5 mg BID (IR 0.07 [95% CI 0.03 to 0.16]) and 13 events occurring in patients receiving an average tofacitinib dose of 10 mg BID (IR 0.09 [95% CI 0.05 to 0.15]).
Gastrointestinal perforations
Gastrointestinal (GI) perforations were reported in 28 (0.4%) patients with an IR (95% CI) of 0.1 (0.1 to 0.2). One additional patient reported a GI perforation outside the 28-day risk period (not included in the IR calculation). IRs for GI perforations with average doses of 5 and 10 mg BID were similar. IRs remained stable with increasing tofacitinib exposure (online supplemental figure 2). In the majority of cases, GI perforations occurred in the lower GI tract (n=22), and all except two occurred in patients with underlying risk factors. Twenty-six patients received concomitant therapy with one or more non-steroidal anti-inflammatory drugs (NSAIDs) or glucocorticoids. Thirteen patients had at least one of the following underlying relevant diseases and/or surgeries: history of gastric perforation with peritonitis, diverticulitis, irritable bowel syndrome, colon polyp, gastroenteritis or gastric ulcer; current event of spastic colon, gastritis, gastroduodenitis or diverticulitis; history of cholecystectomy and/or appendectomy with or without gastric ulcer.
Thromboembolic events
Venous thromboembolism (ie, DVT and/or PE) was reported in 59 (0.8%) patients with an IR (95% CI) of 0.3 (0.2 to 0.3) (table 3). DVT was reported in 36 (0.5%) patients (IR 0.2 [95% CI 0.1 to 0.2]) and PE in 28 (0.4%) patients (IR 0.1 [95% CI 0.1 to 0.2]). Seventeen patients with DVT events (47.2%) and 24 patients with PE events (85.7%) were hospitalised. IR analysis of DVT or PE by 6-month intervals showed that they were generally consistent over time with the overall IR (online supplemental figure 3). Arterial thromboembolism was reported in 84 (1.2%) patients (IR 0.4 [95% CI 0.3 to 0.5]). IRs for all thromboembolic events were similar for both tofacitinib doses in this patient population (table 3). These events were not adjudicated.
Major adverse cardiovascular events
Adjudicated MACE, including myocardial infarction, stroke and/or CV death, was reported in 85 (1.3%) patients (IR [95% CI] of 0.4 [0.3 to 0.5]). MACE IRs were similar for both tofacitinib doses. The IR analysis by 6-month intervals demonstrated that IRs were generally stable with longer tofacitinib exposure (online supplemental figure 4). The most common cardiac disorders reported as SAEs were atrial fibrillation (n=27), myocardial infarction (n=24) and coronary artery disease (n=17).
Laboratory variables of interest
Low-density lipoprotein and high-density lipoprotein cholesterol remained generally stable with tofacitinib treatment over time (online supplemental figure 5).
During the index studies, 10.2% of patients treated with tofacitinib were receiving a statin at baseline, which increased to 15.9% by Month 24 (data truncated at study end in studies <24 months; 14.3% in patients receiving tofacitinib 5 mg BID and 17.9% in patients receiving tofacitinib 10 mg BID). Upon entry into the LTE studies, 12.9% of patients were taking statins, which gradually increased to 25.0% by Months 90–96 (25.1% in patients receiving tofacitinib 5 mg BID and 24.2% in patients receiving tofacitinib 10 mg BID). The use of concomitant statins was at the investigator’s discretion and the specific reason for patients initiating statins was not reported.
Overall, 878 (12.4%) patients had hypertension (IR 4.2 [95% CI 3.9 to 4.5]) and 209 (3.0%) patients had hyperlipidaemia (IR 0.9 [95% CI 0.8 to 1.1]).
Persistent serum creatine kinase elevations were infrequent; these are summarised in the online supplemental results.
DISCUSSION
This long-term integrated safety summary of tofacitinib with up to 9.5 years of follow-up in >7000 patients with RA worldwide, with a combined tofacitinib exposure of 22 875 PY, represents the largest clinical dataset for a JAK inhibitor in RA to date. This 2017 analysis includes cumulative safety data from completed studies in the RA clinical development programme and captures the maximum level of patient exposure to tofacitinib from the main LTE study, ORAL Sequel.
Overall, the crude 2017 IRs of safety events of interest, discontinuations due to AEs and mortality were consistent with those reported in the previous integrated safety analysis of 19 406 PY of tofacitinib exposure and 8.5 years of follow-up,23 and with those observed in the individual phase II,4–8 phase III9–14 and phase IIIb/IV15 RCTs and LTE studies.16–18 IRs for safety events of interest were <0.5/100 PY, with the exception of HZ (all), SIEs, malignancies (excluding NMSC) and NMSC. The crude IRs for safety events of interest were comparable between the two tofacitinib average dosing groups. IRs for SIEs, HZ, OIs (excluding TB), GI perforations, DVT, PE, MACE, malignancies (excluding NMSC) and NMSC remained generally stable with longer exposure to tofacitinib over time. The Cox regression analyses found that increasing tofacitinib doses (based on time-varying dosing in units of 5 mg) could have a higher relative risk of SIEs, HZ and OIs (excluding TB) (HR 1.3, 1.4 and 2.4, respectively).
CV events and SIEs were the most common causes of death; this updated analysis reported that respiratory disorders were the next most common cause of death, whereas the prior integrated safety analysis reported that malignancies were the next most common cause of death.23 The reason for this difference is that the IR calculation used in this study was based on the number of patients with incident events during the time between the first and last dose plus 28 days, which was the clinical trial observation period; the previous integrated safety summary of tofacitinib included events, but not person time, outside the observation period.23
Infections and infestations are typically the most common AEs by SOC reported for tofacitinib.4–18 23 26 Rates of SIEs previously reported in tofacitinib-treated patients with RA were generally comparable with those for patients treated with bDMARDs, baricitinib and upadacitinib.27–31 In this analysis, the most common types of SIEs were pneumonia, HZ, urinary tract infection and cellulitis, which is consistent with the prior analysis23 and those reported for baricitinib and upadacitinib.27 32 Notably, there was an increased risk of SIEs with lymphopenia <500 cells/µL (HR 2.4), which was observed previously,23 and also with lymphopenia <1000 cells/µL (HR 1.3). As noted in the Results section, when stratified by lymphocyte count, the IRs for SIEs were numerically higher for lymphopenia <500 cells/µL versus lymphopenia ≥500–<1000 cells/µL. When assessing the benefit:risk profile of tofacitinib in patients with lymphocyte counts between 500 cells/µL and 1000 cells/µL, consideration is warranted for the magnitude of increase in SIEs and the relatively low frequency of prior confirmed lymphopenia (<500 cells/µL, 76/7061 patients; ≥500–<1000 cells/µL, 1835/7061 patients; note that categories are not mutually exclusive and some patients are included in more than one), as well as the contribution of other risk factors for SIEs in patients that may be more prevalent and have a greater relative impact on risk, such as older age and diabetes.
These results were confirmed in a recent post hoc analysis of combined data from RA tofacitinib phase III and LTE studies, which recommended absolute lymphocyte count monitoring in patients with RA since rates of SIEs are recognised to be increased with lymphopenia.33 34 This is consistent with the requirement in the prescribing information for monitoring of lymphocyte counts at baseline and every 3 months thereafter during tofacitinib therapy.19
The risk of HZ is elevated in patients with RA,35 with further increases in patients treated with JAK inhibitors.23 36–38 Rates of HZ were generally similar to the published rates for baricitinib and upadacitinib,29–31 38 and higher than rates reported for bDMARDs.21 The majority of HZ cases were non-serious, with a low proportion of patients developing disseminated or multi-dermatomal HZ, and were clinically manageable. Results were consistent with previous reports that older patients and those from Japan or Korea had a higher risk for HZ.23 35 The contribution of tofacitinib to an increased risk of HZ in RA is complicated by the fact that over half of patients in the index studies were taking concomitant glucocorticoid therapy. Recent reports suggest that concomitant treatment with glucocorticoids with or without csDMARDs further increases the risk of HZ in patients with RA using tofacitinib39 40; glucocorticoid use was also identified as a risk factor in our multivariate analysis within the present study. A previous analysis of data from the tofacitinib RA clinical development programme demonstrated a trend for numerically higher IR of HZ when tofacitinib was administered with concomitant csDMARDs.39 Consistent with this and other reports, concomitant MTX did not significantly increase the risk of HZ in our analysis.39 40 Lymphopenia was not found to be a significant risk factor for HZ, which is consistent with the previous report.23
The risk of TB and other OIs in patients with RA compared with the general population is increased further in patients treated with bDMARDs41–43 and tofacitinib.44 In our analysis, rates of OIs (excluding TB) were similar to those reported for bDMARDs.45–49 However, between-study comparison of overall rates of OIs with other studies is difficult due to heterogeneous study designs and differences in the definition of OIs.44 Similar to the prior integrated safety analysis of tofacitinib, risk factors for OIs (excluding TB) included higher age, geographical region and increased tofacitinib dose.23 COPD was identified as an additional potential risk factor for OIs (excluding TB) in this analysis. The IR of TB in this study was consistent with previous reports of tofacitinib,17 23 44 with the majority of cases occurring in geographical regions with a high prevalence of TB per prior research, indicating that risk of TB with tofacitinib treatment directly varies with background TB prevalence.44 IR data for TB reported here are comparable with those reported in clinical trials of bDMARDs,50 51 baricitinib and upadacitinib.27 30 31 52 IRs of interstitial lung disease are reported in a separate publication.53
Patients with RA are at a higher risk of developing lung and lymphoma malignancies compared with the general population,54 and continued long-term monitoring of immunomodulatory treatments, such as tofacitinib, is necessary to evaluate any potential malignancy risk.25 There were 12 lymphoma events reported in the main analysis. Of these, 11/12 events were classified as non-Hodgkin lymphoma, and the majority (11/12) occurred in patients receiving an average tofacitinib dose of 10 mg BID. Outside the 28-day risk period window, a total of 19 lymphoma events were reported, 6 events (IR 0.07 [95% CI 0.03 to 0.16]) and 13 events (IR 0.09 [95% CI 0.05 to 0.15]) in patients receiving an average tofacitinib dose of 5 and 10 mg BID, respectively. These results are similar to a previous analysis of lymphoma incidence in the tofacitinib RA clinical development programme, where six lymphoma events were reported in patients receiving an average tofacitinib dose of 5 mg BID, compared with 13 events in those receiving an average tofacitinib dose of 10 mg BID; the majority (17/19 events) were classified as non-Hodgkin lymphoma.55 The risk of malignancies with tofacitinib in this analysis was similar to that reported previously.17 23 It should be noted that estimation beyond 78 months was less precise due to small patient numbers and limited PY of exposure. The IRs and SIRs of malignancies (excluding NMSC) and IRs for NMSC reported here are within a range similar to those reported in RA populations treated with bDMARDs,45 47 56–59 baricitinib and upadacitinib.27 29 Consistent with other studies,59 60 SIRs using GLOBOCAN were higher than those using SEER, potentially due to differences in cancer-screening practices or surveillance systems. However, the need for caution remains over the long-term risk of cancer with any immunomodulator.60
GI perforations are a known risk in patients with RA, and treatment with NSAIDs or glucocorticoids elevates this risk further.61 The IR of GI perforations observed here for tofacitinib was slightly lower than the published rate for tocilizumab,62 slightly higher than the rates reported in the integrated safety studies of certolizumab pegol60 and baricitinib,27 and comparable with the rate reported for upadacitinib.52 However, these variations may be influenced by different exclusion criteria regarding GI medical history between clinical trials.
There is an increased risk of thromboembolic events in patients with RA compared with the general population,63 64 and this risk is increased further with JAK inhibitor treatment.22 27 In contrast to findings in an ad hoc safety analysis of ongoing Study A3921133 described previously in this paper, an increased risk of thromboembolic events was not evident in this 2017 analysis of tofacitinib. A post hoc analysis performed using combined data from phase II/III studies from the RA clinical development programme, which included 5368 patients (representing 4440 PY), did not find a link between tofacitinib treatment and DVT or PE events.65 However, based on the information from Study A3921133, and consideration of information pertaining to PE for other JAK inhibitors, venous thromboembolic events (DVT/PE) are considered an important identified risk for treatment with tofacitinib. The current integrated safety analysis included all patients meeting individual study inclusion/exclusion criteria, irrespective of potential CV risk factors (ie, patients were not required to be ≥50 years of age or have ≥1 CV risk factor) and did not include Study A3921133.
Based on pooled data from RCTs and LTEs, the IR of MACE reported with tofacitinib appears similar to that reported with bDMARDs, such as certolizumab pegol (0.62/100 PY)60 and tocilizumab (0.34/100 PY),66 and with baricitinib (0.8/100 PY)67 and upadacitinib (0.6/100 PY).29 Head-to-head studies, such as Study A3921133 described earlier, are necessary to identify any differences in CV risk between treatments.
Older patients had an increased risk of SIEs, HZ and OIs. The safety of tofacitinib in patients aged <65 years versus ≥65 years has been evaluated previously in post hoc analyses of data pooled from clinical studies. Incidence of SAEs and discontinuations due to AEs were generally higher in older versus younger patients in a post hoc analysis of data from phase III and LTE studies.68 In addition, a recent analysis of tofacitinib studies that included an adalimumab control/comparator arm reported that IRs of overall infection events and SIEs were higher with tofacitinib 5 mg BID, tofacitinib 10 mg BID and adalimumab in patients aged ≥65 years versus <65 years. The risk of SIEs or overall infection events was similar for tofacitinib and adalimumab, with the exception of a numerically higher rate of SIEs with tofacitinib 10 mg BID versus adalimumab in patients aged ≥65 years.69 Duration of RA is an important factor in predicting response to treatment; however, duration of RA was not a significant risk factor for SIEs, HZ or OIs in the present analysis. A previous post hoc analysis of patients with RA duration of <1 year versus ≥1 year receiving tofacitinib monotherapy in the phase III ORAL Start study found no difference in safety profiles with longer versus shorter RA duration.70 In addition, a post hoc analysis of data from patients receiving tofacitinib in the phase IIIb/IV ORAL Strategy study found that rates of AEs were generally similar in those with early RA (duration ≤2 years) compared with established RA (duration >2 years).71 Use of glucocorticoids at baseline was associated with a higher risk of SIEs and HZ. A previous post hoc analysis of data pooled from phase I, phase II, phase III and LTE studies of tofacitinib also found that risk of HZ was higher in those receiving tofacitinib with glucocorticoids at baseline, versus tofacitinib monotherapy.39 However, a separate post hoc analysis of data from a phase IIIb/IV study found no difference in rates of AEs with glucocorticoid use; therefore, the effect of glucocorticoids on safety endpoints in patients with RA receiving tofacitinib may require further study.72
As reported previously, there were limitations to this integrated analysis.23 Although analysis of crude IRs did not account for the imbalance and potential dose switching between 5 mg and 10 mg BID doses and the different dosing requirements for different geographical regions, the majority (76.4%) of patients remained on their start dose in the LTE studies. As a pertinent limitation to the dose-related analyses, the use of average daily dose estimated at the end of the study, rather than in a time-varying fashion, may have attenuated any safety differences between tofacitinib doses. Throughout the course of the trial, 23.6% of the patients changed dose at any time. Additionally, while the average dose approach categorises patients into either tofacitinib 5 or 10 mg BID, the Cox regression model takes into account the actual dose from the index studies (ie, some patients were treated with >10 mg BID and others with <5 mg BID). Therefore, it is not possible to reconcile the results from the Cox regression model with the crude IRs since the method used to classify dose was dissimilar between the two approaches. Caution should also be taken when interpreting results for patients with the shortest and longest tofacitinib exposure due to differences in patient numbers, which are fewer in later months, and reporting AEs as counts or rates assume a constant hazard (risk) over time. Another limitation was that changes in rates of SAEs over time and recurring SAEs could not be analysed because patients who developed certain SAEs were discontinued and censored at the time of first event. This restricts the patient population to those in which tofacitinib was well tolerated, restricting full benefit:risk profile evaluation, although this also reflects clinical practice where patients experiencing SAEs would discontinue therapy. Our analysis found that IRs for AEs of interest remained stable with longer tofacitinib exposure. It should be noted that this is common in any long-term safety study of treatments for RA, or other treatments, and is likely partially due to the survival bias (ie, depletion of susceptibles), as those who tolerate therapy without AEs are more likely to continue such therapy.73 Another potential limitation is that, with the exception of assessment of lymphoma events, this analysis used an updated IR calculation that classified exposure based on treatment received plus 28 days after discontinuation versus the method used in the previous analysis that included events, but not person time, outside the observation period,23 which must be considered when making comparisons. While this means that events recorded beyond the observation period were not included in the calculation of IRs, as they had been in the previous analysis, this updated approach is a more systematic method of event recording versus the previous method as it ensures no differential follow-up for case ascertainment for any patients at risk.
Comparisons with placebo were not included because treatment duration with placebo was short, thus limiting the PY of placebo exposure. The analysis is also limited by the difficulty in accurately assessing the frequency of events of long latency, as they may not have occurred during the study period.
CONCLUSIONS
This 2017 long-term integrated safety analysis represents the largest and longest clinical dataset for a JAK inhibitor for RA and captures the maximum level of patient exposure to tofacitinib from the main LTE study, ORAL Sequel, which is now completed. Rates of important safety events, including infections and malignancies, were stable across data cuts. In this analysis, with the exception of higher HZ rates than those reported for bDMARDs, particularly among patients from Japan and Korea, the rates of safety events were generally similar to those reported for bDMARDs and other JAK inhibitors used to treat RA.
Studies collecting patient safety data on tofacitinib beyond clinical trials are currently ongoing. For example, the prospective study using data from the US Corrona RA registry is evaluating tofacitinib safety in a US real-world population,74 while the British Society for Rheumatology Biologics Register for Rheumatoid Arthritis, the Antirheumatic Therapies in Sweden and the Rheumatoide Arthritis: Beobachtung der Biologika‐Therapie registers are collecting long-term data on real-world use of tofacitinib and other drugs used to treat RA in Europe.
Acknowledgments
The authors would like to thank Aditya Patel (Syneos Health) and Arti Kanujia (Covance) for statistical support. Medical writing support, under the guidance of the authors, was provided by Jennifer Higginson, PhD, CMC Connect, McCann Health Medical Communications and was funded by Pfizer Inc, New York, NY, USA, in accordance with Good Publication Practice (GPP3) guidelines (Ann Intern Med 2015;163:461–64).
REFERENCES
Footnotes
Correction notice In Figure 3 of the originally published version of the manuscript, the incidence rates for all tofacitinib does from months 0-6 >78 were incorrectly designated for panels (A) malignancies (excluding NMSC) and (B) NMSC. The data have now been switched to the correct designation.
Contributors Involved in the conception and design of the study/analyses: SBC, YT, XM, JRC, EBL, PN, KLW, CC-S, LW, CC, AS, AM and JW. Involved in patient recruitment, study monitoring, and/or data acquisition (conducted the experiment): YT, LW and EBL. Performed the data and statistical analyses: LW, CC, KK, PB, AS and AM. All authors were involved in data interpretation and manuscript drafting, reviewing and development. The views and opinions expressed within this manuscript are those of all authors and do not necessarily represent those of the sponsor.
Funding This study was sponsored by Pfizer Inc.
Competing interests SBC has received grant/research support from AbbVie, Amgen, Astellas, Bristol-Myers Squibb, Eli Lilly, Genentech, Gilead, Janssen, Novartis, Pfizer Inc, Roche and Sandoz; and consultancy fees from AbbVie, Amgen, Astellas, Bristol-Myers Squibb, Eli Lilly, Genentech, Gilead, Janssen, Novartis, Pfizer Inc, Roche and Sandoz. YT has received grant/research support from AbbVie, Asahi Kasei, Astellas, Bristol-Myers Squibb, Chugai, Daiichi Sankyo, Eisai, Eli Lilly, Gilead, GlaxoSmithKline, Janssen, Mitsubishi Tanabe, Novartis, Pfizer Inc, Sanofi and YL Biologics; and speaker fees and/or honoraria from AbbVie, Astellas, Bristol-Myers Squibb, Chugai, Daiichi Sankyo, Eisai, Eli Lilly, Janssen, Mitsubishi Tanabe, Novartis, Pfizer Inc, Takeda, Teijin and YL Biologics. XM has received consultancy fees from Bristol-Myers Squibb, Gilead, GlaxoSmithKline, Janssen, Pfizer Inc, Samsung and UCB. JRC has received grant/research support from Amgen, Corrona, Crescendo Bio and Pfizer Inc; and consultancy fees from AbbVie, Amgen, Bristol-Myers Squibb, Corrona, Eli Lilly, Janssen, Myriad, Pfizer Inc, Roche/Genentech and UCB. EBL has received consultancy fees from Pfizer Inc. PN has received grant/research support and consultancy fees from, and is part of the speakers’ bureau for, AbbVie, Bristol-Myers Squibb, Eli Lilly, Janssen, Novartis, Pfizer Inc, Roche, Sanofi and UCB. KLW has received grant/research support and consultancy fees from AbbVie, Bristol-Myers Squibb, Eli Lilly, Gilead, Pfizer Inc, Roche and UCB. CC-S has received grant/research support from AbbVie, Bristol-Myers Squibb and Pfizer Inc; and consultancy fees from Gilead, Pfizer Inc and Regeneron-Sanofi. LW, CC, KK, PB, AS and AM are employees and shareholders of Pfizer Inc. JW has received consultancy fees from, and is on the speaker’s bureau for, Pfizer Inc.
Patient consent for publication All patients provided written informed consent.
Ethics approval Studies were conducted in accordance with the Declaration of Helsinki and the Good Clinical Practice Guidelines, along with applicable local country regulations and laws. The study protocols were approved by the Institutional Review Boards and/or Independent Ethics Committee at each centre.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Upon request, and subject to certain criteria, conditions and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de-identified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines, and medical devices (1) for indications that have been approved in the USA and/or EU, or (2) in programmes that have been terminated (ie, development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de-identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.