Article Text

Abstract
Objectives Patients with lupus membranous nephropathy (LMN) are at risk for prolonged proteinuria and progressive chronic kidney disease. There are no proven effective treatments for LMN, and controlled trials are lacking. This trial assessed the preferential Janus kinase 1 (JAK1) inhibitor filgotinib and the spleen tyrosine kinase inhibitor lanraplenib in patients with LMN.
Methods This was a phase II, randomised, double-blind trial conducted at 15 centres in the USA to evaluate the safety and efficacy of filgotinib or lanraplenib for the treatment of LMN. Eligible patients were randomised 1:1 to receive either filgotinib or lanraplenib in a blinded fashion for up to 52 weeks. The primary endpoint was the per cent change in 24-hour urine protein from baseline to week 16.
Results Nine patients were randomised to receive filgotinib (n=5) or lanraplenib (n=4). Four patients in the filgotinib group and one patient in the lanraplenib group completed week 16. There was a median reduction of 50.7% in 24-hour urine protein after 16 weeks of treatment with filgotinib (n=4), and the median Systemic Lupus Erythematosus Disease Activity Index from the Safety of Estrogens in Lupus National Assessment score remained stable. Filgotinib treatment was well tolerated. Limited conclusions can be drawn about treatment with lanraplenib.
Conclusion The number of patients treated in this study was small, and only limited conclusions can be drawn. There may be a therapeutic benefit with filgotinib treatment, which may support future investigations with filgotinib or other JAK inhibitors in patients with LMN.
Trial registration number NCT03285711.
- lupus erythematosus
- systemic
- lupus nephritis
- therapeutics
Data availability statement
No data are available.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
There are no clearly effective therapies for lupus membranous nephropathy (LMN), and controlled trials in patients with LMN are lacking.
What does this study add?
Our study demonstrates that 16 weeks of treatment with the Janus kinase 1 (JAK1) inhibitor filgotinib reduced proteinuria in a small number of patients with LMN.
No benefit was seen in patients with LMN after treatment with the spleen tyrosine kinase inhibitor lanraplenib, and a high dropout rate was observed.
How might this impact on clinical practice?
Limited conclusions can be drawn from this small study, but filgotinib treatment may provide a therapeutic benefit in patients with LMN.
Future studies are needed to more definitively assess the benefit of JAK inhibition in LMN.
Introduction
Systemic lupus erythematosus (SLE) is a chronic, systemic, multiorgan autoimmune disease. Depending on the population studied, up to 50% of patients with SLE develop renal involvement, which can vary from asymptomatic haematuria to nephrotic syndrome or rapidly progressive glomerulonephritis.1 Lupus membranous nephropathy (LMN) occurs in 10%–20% of patients with lupus nephritis, and it can result in protracted nephrotic syndrome and an increased risk of developing end-stage renal disease.1 2 In addition, patients with LMN are predisposed to hypercholesterolaemia, hypertension, accelerated atherosclerosis, hypercoagulability and an increased risk of infection.3
There are limited studies investigating the benefit of conventional synthetic disease modifying antirheumatic drugs (DMARDs), biological DMARDs (bDMARDs) or targeted synthetic DMARDs in the treatment of LMN.4–12 In particular, randomised, controlled trials are lacking. Prior studies support the use of prednisone, azathioprine, mycophenolate mofetil, cyclophosphamide, cyclosporine, chlorambucil and plasmapheresis.4–11 13 14 However, these investigations have been small, have used varied outcome measures and have not always reported dose and duration of therapy. Currently, the first-line recommended induction regimen for LMN involves high-dose glucocorticoids plus mycophenolate mofetil.15 16 However, these recommendations are based on the limited available studies, and a consensus regarding optimal second-line treatment does not exist.
Filgotinib is a potent small molecule inhibitor of the Janus kinase family of enzymes (JAKs), with preferential selectivity for JAK1.17 Filgotinib, much like other JAK inhibitors (JAKi), has shown efficacy in the treatment of rheumatoid arthritis (RA).18–21 In a phase III study of patients with moderately to severely active RA and inadequate response to one or more prior bDMARDs, 66% of patients receiving filgotinib 200 mg/day achieved 20% improvement in the American College of Rheumatology criteria (ACR20) at week 12, compared with 31.1% of patients receiving placebo.22 In addition, a study for the treatment of diabetic kidney disease (DKD) with baricitinib, a JAK1/2 selective inhibitor, indicated a dose-related reduction in albuminuria and inflammatory biomarkers involved in DKD progression.23 JAK1 is involved in the signalling of cytokines that are believed to play critical roles in SLE pathogenesis, such as interferon alpha, interleukin (IL)-10, IL-21 and IL-6.24–33
Lanraplenib (previously known as GS-9876) is a selective and potent ATP-competitive inhibitor of spleen tyrosine kinase (SYK).34 The SYK pathway plays an important role in inflammatory disease, primarily through the activation, survival and migration of B cells and through Fc-gamma receptor-mediated myeloid cell activation.35–38 SYK activation leads to downstream signalling through the phosphatidylinositol 3-kinase, Bruton’s tyrosine kinase and mitogen-activated protein kinase pathways.
Given the toxicity and variable efficacy of current immunosuppressive medications used in the treatment of LMN, there is an unmet need for safer, more effective therapies. Filgotinib and lanraplenib both serve as rational treatment options, and this study was designed to assess their safety and efficacy in the treatment of patients with LMN.
Methods
Patients
This Food and Drug Administration—approved phase II, multicentre, randomised, double-blind trial was conducted from September 2017 through February 2020 at 15 centres in the USA. Eligible patients were 18–75 years of age with a positive antinuclear antibody (>1:40) and/or positive anti-dsDNA antibody. Inclusion criteria included a kidney biopsy performed within 18 months prior to screening that demonstrated a histological diagnosis of LMN based on the International Society of Nephrology and the Renal Pathology Society 2003 classification of lupus nephritis, with either Class V alone, or Class V in combination with Class II. In addition, eligible patients had a urine protein excretion ≥1.5 g/day based on a 24-hour urine sample, an estimated glomerular filtration rate (eGFRMDRD) ≥60 mg/min/1.73 m2 based on the Modification of Diet in Renal Disease (MDRD) formulation, and prior treatment for LMN with at least one immunosuppressive therapy (mycophenolate mofetil, azathioprine, tacrolimus, cyclosporine, cyclophosphamide or chlorambucil) for at least six consecutive months within 1 year before screening. Oral glucocorticoids were allowed as long as the dose was ≤20 mg/day of prednisone or equivalent and remained stable through week 16 of the study. Patients were also permitted to continue hydroxychloroquine at a stable dose. Treatment with an ACE inhibitor or angiotensin II receptor blocker, or documented intolerance, was required. All immunosuppressive agents other than glucocorticoids, hydroxychloroquine, azathioprine (up to a maximum of 3 mg/kg body weight/day or 300 mg/day), and methotrexate (≤20 mg per week) were withdrawn at least 28 days prior to day 1. Patients were excluded if they had received previous treatment with a JAKi within 3 months of day 1 or rituximab or other B cell depleting agent within 6 months of day 1. In order to enhance recruitment, in March 2018 the protocol was amended to extend the window for kidney biopsy to 36 months prior to screening. In addition, the eGFR inclusion criteria was modified to allow all patients with an eGFRMDRD≥40 mL/min/1.73 m2. The duration of prior immunosuppressive treatment was changed to the discretion of the investigator, and additional treatments such as methotrexate, leflunomide and moderate-dose to high-dose glucocorticoids were permitted.
Treatment protocol
Subjects were randomised in a 1:1 ratio to receive once daily oral doses of either filgotinib 200 mg (plus placebo to match (PTM) lanraplenib) or lanraplenib 30 mg (plus PTM filgotinib). Randomisation was stratified by prior treatment with cyclophosphamide. All subjects who achieved a ≥35% reduction in urinary protein excretion at week 16 compared with baseline continued to receive their assigned blinded study treatment for an additional 16 weeks. The average of two spot urine protein to creatinine ratios (UPCR) from two consecutive morning voids prior to the week 16 visit were used to determine response for subsequent study treatment. For those subjects who did not achieve a ≥35% reduction in urinary protein excretion, their study treatment was switched for the next 16 weeks in a blinded fashion (ie, those on filgotinib+PTM lanraplenib switched to lanraplenib+PTM filgotinib, while those on lanraplenib+PTM filgotinib switched to filgotinib+PTM lanraplenib). After 32 weeks of blinded treatment, those who had a ≥35% reduction in urinary protein excretion from day 1 (for subjects who remained on the randomised study treatment after week 16) or from week 16 (for subjects who switched treatment at week 16) continued their assigned blinded treatment for an additional 20 weeks in the Extended Blinded Treatment Phase. Subjects who did not achieve a ≥35% reduction in urinary protein excretion at week 32 compared with baseline were allowed to continue whichever study treatment led to the greatest reduction in urinary protein excretion at the subject’s and investigator’s discretion. The use of rescue therapy (a new, or increased dose of an existing, immunosuppressant agent, including glucocorticoids) required discontinuation of the study treatment.
Study endpoints
The primary efficacy endpoint was the per cent change in urine protein from baseline to week 16 assessed during a 24-hour urine collection. Secondary endpoints comparing week 16 to baseline included: the absolute change in 24-hour urine protein, the change in eGFR, the change in the spot UPCR, the change in the UPCR based on the 24-hour urine collection, the proportion of subjects achieving partial remission (defined as urine protein excretion <3 g/day and urine protein excretion decrease by ≥50% among subjects with baseline nephrotic range proteinuria (urine protein excretion ≥3 g/day); or urine protein excretion decrease by ≥50% among subjects with subnephrotic range proteinuria (urine protein excretion <3 g/day)), the proportion of subjects achieving complete remission (defined as urine protein excretion below 0.5 g/day with no haematuria). Additional exploratory endpoints (comparing week 16 to baseline): the change in complement components, the change in anti-dsDNA antibody levels and the change in disease activity scores such as the Systemic Lupus Erythematosus Disease Activity Index from the Safety of Estrogens in Lupus National Assessment study (SELENA-SLEDAI), the British Isles Lupus Assessment Group index, Patient Global Assessment of disease activity score (PGA) and Physician Global Assessment of disease activity score (PhGA). Safety was assessed during the study through the reporting of adverse events (AEs) and lab abnormalities.
Statistical analysis
The study planned to enrol 32 subjects (16 per arm). This original sample size was chosen based on the assumption that 16 subjects per treatment group (32 subjects total), would provide 80% power to detect a 35% reduction from baseline in proteinuria at week 16 with an SD of 50% and a two-sided 0.05 significance level.
However, only nine subjects were enrolled. Therefore, inferential analyses were not performed, and all statistical analyses are descriptive.
Baseline characteristics are described for all patients enrolled in the study. For the primary endpoint, the per cent change from baseline to week 16 in 24-hour urine protein excretion was reported for each treatment group, including only those patients that reached week 16.
All continuous endpoints were summarised using the median by treatment group given the small sample size. All categorical endpoints were summarised by the number and percentage of subjects who met the endpoint definition.
Safety analyses included all data from baseline through week 52.
Results
Study population
This trial, which was originally designed to include 32 subjects, was not completed due to enrollment challenges. It was ultimately limited to nine patients who were randomised to receive filgotinib (five patients) or lanraplenib (four patients) (figure 1). Of the nine subjects who were randomised, three subjects (33.3%) completed the study. Of the six subjects (66.7%) who prematurely discontinued the study, three discontinued due to an AE, two discontinued due to lack of efficacy and one discontinued due to a protocol violation.
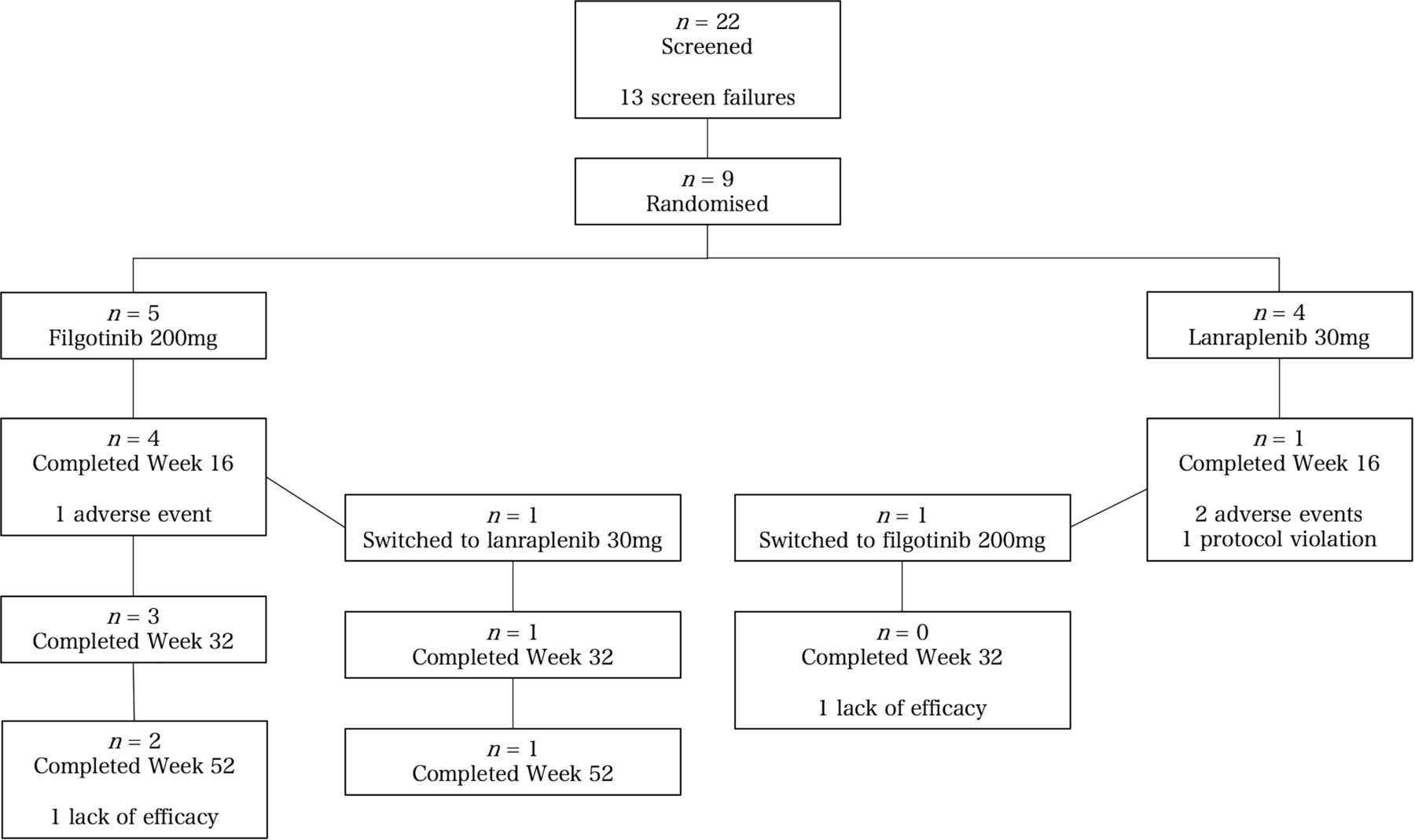
Disposition of subjects.
In the filgotinib group, four patients completed week 16 and three patients completed week 52 (figure 1). One patient in the filgotinib group switched to lanraplenib at week 16. In the lanraplenib group, one patient completed week 16 and no patients completed the study.
In the filgotinib and lanraplenib groups, patients were a median of 28 years and 36 years of age, 80% women and 75% women, and 60% black and 100% black, respectively (table 1). The median baseline 24-hour urine protein for all patients was 2.9 g (2.4 g in the filgotinib group and 4.8 g in the lanraplenib group), and the median baseline spot UPCR was 2.1 mg/mg (1.9 mg/mg in the filgotinib group and 4.2 mg/mg in the lanraplenib group). The median baseline eGFR for the combined groups was 101.0 mL/min/1.73 m2 (100.4 mL/min/1.73 m2 in the filgotinib group and 119.0 mL/min/1.73 m2 in the lanraplenib group). Two patients in each group had previously received cyclophosphamide, three patients in each group had previously received mycophenolate mofetil, and one patient in the lanraplenib group had previously received azathioprine. Glucocorticoids were concomitantly used in four patients treated with filgotinib and three patients treated with lanraplenib, with mean doses of 13.1 mg/day and 15.0 mg/day, respectively (table 1).
Demographics and baseline characteristics of patients with lupus membranous nephropathy
Efficacy outcomes
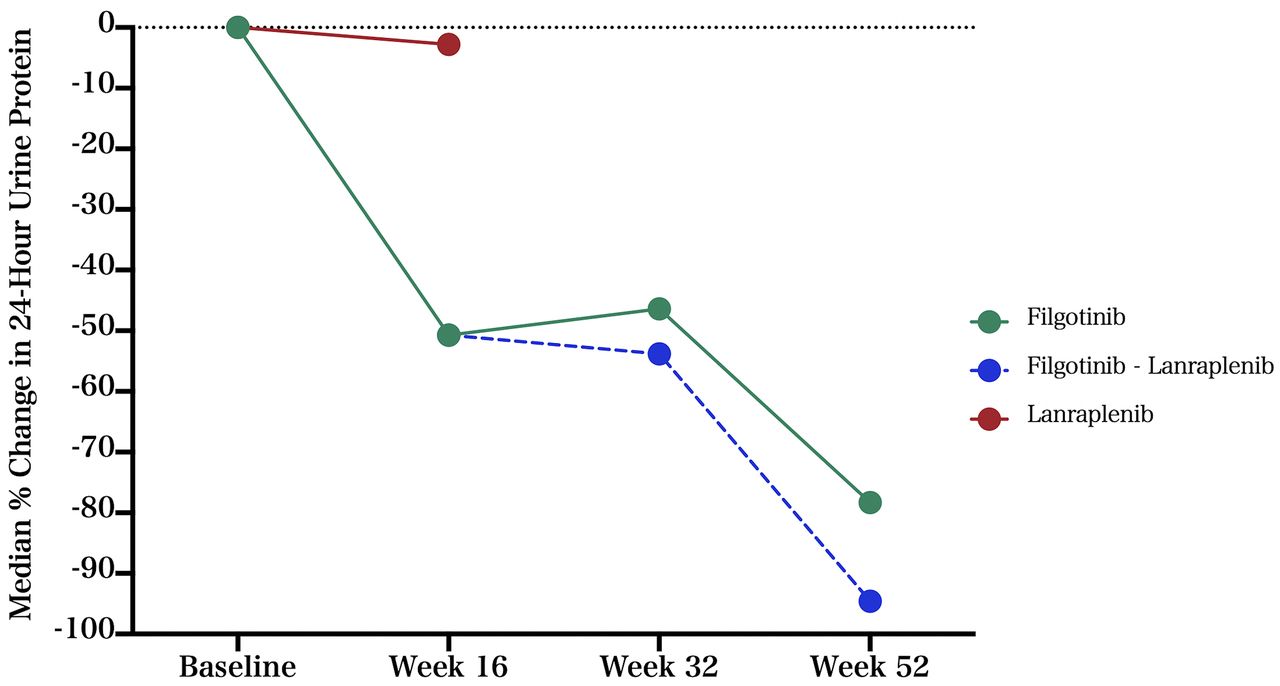
At week 16, the median per cent change from baseline in 24-hour urine protein was −50.7% for the filgotinib group (n=4) and −2.8% for the lanraplenib group (n=1) (table 2 and figure 2).
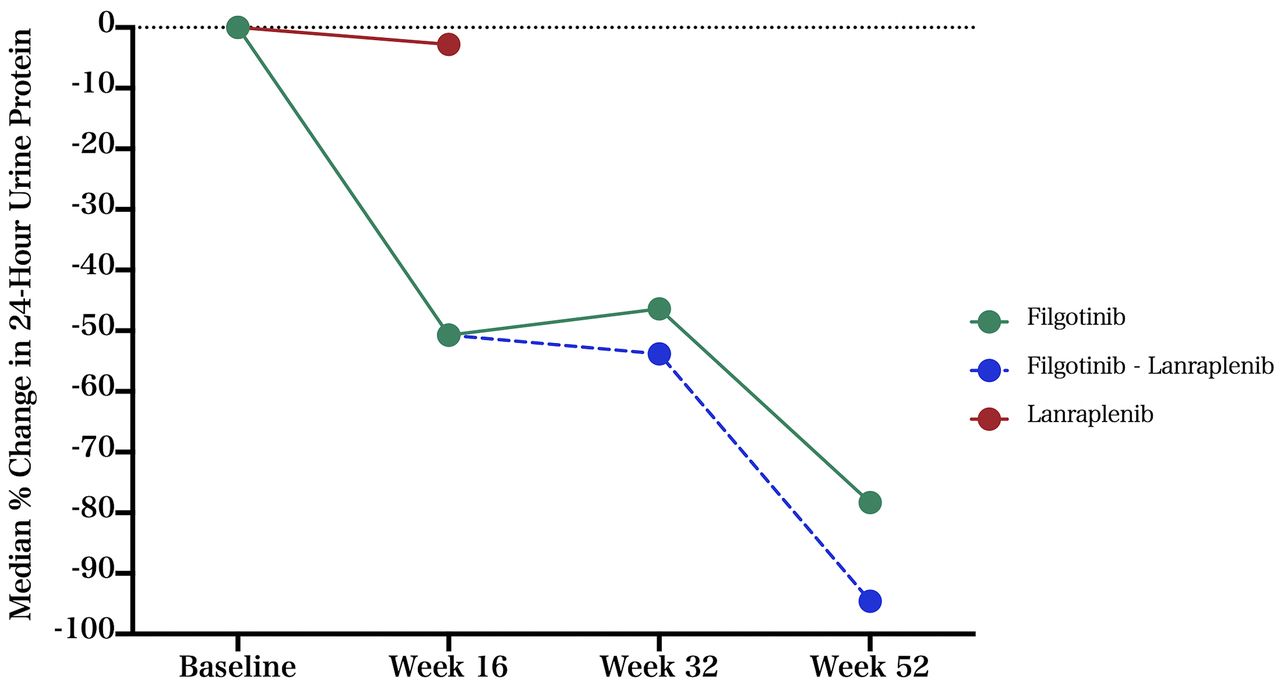
Median per cent change in 24-hour urine protein.
Change from baseline to week 16 in efficacy endpoints
For the four patients in the filgotinib group and the one patient who remained in the lanraplenib group, the median changes from baseline to week 16 were −1.4 g/day and −0.2 g/day in 24-hour urine protein, −0.4 mg/mg and −2.2 mg/mg in the spot UPCR, −0.7 mg/mg and −4.4 mg/mg in the UPCR based on 24-hour urine collection, and −1.2 mL/min/1.73 m2 and −59.4 mL/min/1.73 m2 in eGFR, respectively (table 2 and figure 3). Figure 3 depicts all three urine protein measures on the individual patient level.

{kind=link}
{kind=link}
{kind=link}
Patient-level changes in three different urine protein measures.
At week 16, one of the four patients in the filgotinib arm changed to lanraplenib, as the 12% reduction in the spot UPCR observed did not meet the 35% improvement threshold to remain on filgotinib. Notably, the 24-hour urine collection demonstrated a 70% reduction in protein and the UPCR based on 24-hour urine collection demonstrated a 51% reduction. This patient continued on lanraplenib for the remainder of the study, with a 94.6% reduction in 24-hour urine protein observed at week 52 compared with baseline. Two patients in the filgotinib group remained on filgotinib for the duration of the study, with a median reduction in 24-hour urine protein of 78.3% at week 52 (figure 3 and online supplemental table S1).
Supplemental material
The SELENA-SLEDAI total score remained stable for three of the four patients in the filgotinib group from baseline to week 16 with a median score for those four patients of 7 at both timepoints (online supplemental figure S1). For the two patients in the filgotinib group who reached week 52, the SELENA-SLEDAI score for one patient was 0 at baseline and 0 and week 52 and for one patient improved from 6 at baseline to 2 at week 52 (online supplemental figure S2). The PhGA of disease activity improved by 71.4% and the PGA of disease activity improved by 50.8% for the four patients treated with filgotinib at week 16 (table 2). For the lanraplenib group, the PhGA worsened by 14% and the PGA improved by 33.3% after 16 weeks of treatment. No improvement in anti-dsDNA, C3, or C4 was seen in either group (table 2).
Safety
The majority of subjects in both treatment periods reported at least one AE (table 3). Up to week 16, grade ≥3 AEs were reported in one subject (20%) in the filgotinib group and three subjects (75%) in the lanraplenib group. After week 16, only one subject reported an AE of grade ≥3 (filgotinib/lanraplenib group). Most AEs were not considered related to the study drug. One subject in the lanraplenib arm reported two treatment-emergent Serious Adverse Events (SAEs) and both were not considered to be related to study drug.
Treatment-emergent adverse events for all study participants
Table 3 presents AEs reported during the two treatment periods. Up to week 16, the most common AEs reported in ≥2 subjects were neutropenia (two subjects (40%) in the filgotinib group) and bronchitis (two subjects (50%) in the lanraplenib group). AEs reported after week 16 occurred in ≤1 subject in any treatment group.
Overall, the following grade ≥3 treatment-emergent AEs (TEAEs) were reported: neutropenia, decrease in lymphocyte count, hypercholesterolaemia, hypoalbuminaemia, worsening of SLE and acute kidney injury (one subject each). All TEAEs are listed in online supplemental table S2. From baseline to week 16, one subject (20%) in the filgotinib group and two subjects (50%) in the lanraplenib group prematurely discontinued study drug due to TEAEs (online supplemental table S3). No TEAE leading to study drug discontinuation was reported after week 16. Up to week 16, only one subject (lanraplenib group) reported a grade ≥3 TEAE assessed by the investigator as related to the study drug, which was a decrease in the lymphocyte count. There were no TEAEs grade ≥3 that were assessed as related to the study drug after week 16. The grade 3 laboratory abnormalities that occurred were a decrease in the neutrophil count, a decrease in the lymphocyte count and hypoalbuminaemia.
There were no reports of venous thromboembolism, herpes zoster, malignancy or death during the study.
Discussion
Nine patients with LMN were enrolled and randomised into this study, which is the largest published series of lupus patients treated with filgotinib or lanraplenib to date. In the filgotinib group, 24-hour urine protein was reduced by 50.7% at week 16 compared with baseline for the four patients who remained at week 16. Although the interpretation of these findings is limited, filgotinib appeared to provide meaningful benefit in the small number of patients treated. Only one patient in the lanraplenib group reached week 16, and no significant reduction in 24-hour urine protein was seen. The lanraplenib group had a higher proportion of black patients and a higher baseline 24-hour urine protein compared with the filgotinib group.
Treatment with filgotinib 200 mg/day was generally well tolerated. Given the small number of patients treated with lanraplenib through week 16, limited conclusions can be drawn about its safety. The most common AEs during the study were neutropenia and bronchitis (two patients each). Most AEs reported were grade 1 or 2 in severity. No treatment-related SAEs were reported, and no deaths were reported during the study. Grade 3 laboratory abnormalities included a decrease in the neutrophil count, a decrease in the lymphocyte count and hypoalbuminaemia. No patient had a grade 4 treatment-emergent laboratory abnormality during either treatment period, and no patient met the criteria for liver-related laboratory abnormalities. There were no reports of venous thromboembolism; however, for patients with LMN with heavy nephrotic range proteinuria, this may still be a theoretical concern.
In the filgotinib group, potential benefit was observed with several additional efficacy endpoints as well. At week 16, the PhGA of disease activity improved by 71.4% and the PGA of disease activity improved by 50.8%. The median SELENA-SLEDAI score remained stable during 16 weeks of treatment. No meaningful change in anti-dsDNA, C3 or C4 was observed.
Several challenges were encountered during this study. The primary endpoint of change in urine protein was selected as an objective laboratory test that defines disease severity. However, this study highlights issues surrounding the logistics and reliability of urine protein as an outcome measure. Obtaining two first morning voids followed by 24-hour urine collection requires strict compliance with complex instructions and can be burdensome for patients. In addition to collection materials, patients were given refrigerant packs and a cooler, and they needed to store the first morning voids in the refrigerator until the next study visit. Urine samples were additionally divided into two parts in order to add boric acid tablets to half the samples for preservation purposes. This posed technical challenges for clinical research coordinators. Importantly, this study observed discordance between the 24-hour urine protein, spot UPCR and UPCR based on 24-hour urine. This is in line with several prior studies that documented discordance between the measures, and in contrast to other studies that showed concordance.39–43 The discordance seen in this study resulted in one patient with a good response to filgotinib based on 24-hour urine protein switching to lanraplenib at week 16, perhaps unnecessarily (P4 in figure 3). Future studies that aim to use spot UPCR as part of the treatment response criteria should consider the challenges of such discordance.
This study had several limitations. It was not placebo-controlled, and thus although patients were blinded to their treatment assignment, all patients knew they were receiving one active therapy. The potential bias introduced by this was minimised by selecting an objective laboratory measure as the primary endpoint and by including PTM the alternative treatment not being received. The crossover design may have led to premature designation of treatment failure, however, crossover only occurred for two patients in the study. In addition, this study did not test multiple doses of either treatment, and thus we cannot be certain that the appropriate dose for LMN was chosen. The small sample size and high dropout rate are additional limitations that make it difficult to draw meaningful conclusions from the data. Finally, the high proportion of black patients that were treated in this study may reduce the generalisability of our results to other patient populations.
This study also has several strengths. It is the first prospective study for the treatment of LMN with a JAK inhibitor or SYK inhibitor. Data were collected not only on the response to treatment on proteinuria, but also patient reported outcomes, disease activity indices and additional laboratory values. For the small number of patients who completed the study, the duration was long, with three patients completing 52 weeks of treatment.
LMN is a challenging disease to treat and to study. This clinical trial did not reach its recruitment goal, in part due to small numbers of patients with LMN and in part due to the desire of patients, referring physicians and investigators to continue background medications that were not permitted in the study. Although no clearly effective treatments exist for LMN, patients often derive partial benefit from DMARDs such as mycophenolate mofetil, and we observed a reluctance to stop such medications in order to receive an experimental therapy. Future studies may consider allowing concomitant treatment with mycophenolate mofetil in order to maximise study recruitment and treatment response.
Conclusion
In this randomised, double-blind clinical trial, five patients with LMN were treated with filgotinib and four patients were treated with lanraplenib. A median reduction of 50.7% in 24-hour urine protein was seen after 16 weeks of filgotinib treatment for the four patients who completed week 16. The lanraplenib group had a high dropout rate and no reduction in urine protein was seen. For patients who received filgotinib, the median SELENA-SLEDAI score remained stable through 16 weeks. There was no improvement in anti-dsDNA or complement levels for either group. The number of patients treated in this study was small, and only limited conclusions can be drawn. The potential therapeutic benefit observed with filgotinib treatment may support future investigations with filgotinib or other JAK inhibitors in patients with LMN.
Data availability statement
No data are available.
Ethics statements
Patient consent for publication
Ethics approval
The study protocol was approved by institutional review boards at each study centre, including The Stanford University institutional review board (IRB-42050), and all patients provided written informed consent.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @mbake013
Correction notice This article has been corrected since it first published. The provenance and peer review statement has been included.
Contributors MBaker, UP and MG were involved in the initial design and planning of the study. All authors were involved in the conduct and reporting of the work described in the article. MBaker, UP and MG are responsible for the overall content as guarantors.
Funding MBaker received support for this work from the KL2 component of the Stanford Clinical and Translational Science Award to Spectrum (NIH NCATS KL2 TR 001083). This study was sponsored by Gilead Sciences.
Competing interests MBaker reports fees from Gilead unrelated to this study; MG, HH, OG, UP report employment and stock from Gilead Sciences.
Provenance and peer review Not commissioned; externally peer reviewed.