Article Text

Abstract
Objective Interleukin (IL)-6 is a pleiotropic cytokine involved in the pathophysiology of rheumatoid arthritis (RA). Sirukumab is a human monoclonal antibody that binds to IL-6 with high affinity and specificity.
Methods This long-term extension (LTE) study of the SIRROUND-D and SIRROUND-T studies assessed long-term safety and efficacy of sirukumab in adults with moderate-to-severe RA refractory to conventional disease-modifying antirheumatic drug therapy or antitumor necrosis factor agents. Patients received sirukumab 100 mg subcutaneously (SC) every 2 weeks (q2w) or sirukumab 50 mg SC every 4 weeks (q4w).
Results 1820 patients enrolled in the LTE; median exposure was 2.34 and 2.07 years in sirukumab 50 mg q4w and 100 mg q2w groups, respectively. Adverse events (AEs) occurred in similar proportions between groups, with the exception of major adverse cardiovascular events (MACE), which were more common in the 50 mg q4w versus 100 mg q2w group (2.2% vs 1.0%), and injection-site reactions, more common in the 100 mg q2w group versus 50 mg q4w group (7.5% vs 3.7%). The most common serious AEs were infections (10% of the patients); 32 (1.8%) patients died during the study (primarily from serious infection and MACE). Malignancies were reported in 24 (1.3%) patients. Gastrointestinal perforations, hepatobiliary abnormalities and changes in laboratory parameters were rare. Reductions in RA signs and symptoms and improvements in physical function were maintained throughout the LTE.
Conclusions The safety profile of sirukumab in the LTE remained consistent with that reported in SIRROUND-D and SIRROUND-T and efficacy was maintained.
Trial registration number NCT01856309.
- arthritis
- rheumatoid
- therapeutics
- antirheumatic agents
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Inhibition of interleukin (IL)-6 receptor (IL-6R) is an established, effective and safe approach for treatment of rheumatoid arthritis (RA).
The IL-6 cytokine inhibitor sirukumab has been studied in a pivotal trial programme, and has been shown to be of comparable efficacy and safety to existing IL-6R inhibitors.
What does this study add?
The long-term extension (LTE) study of sirukumab confirms that reductions in RA signs and symptoms and improvements in physical function were maintained.
The safety profile of sirukumab in the LTE remained consistent with that reported in pivotal SIRROUND-D and SIRROUND-T trials.
How might this impact on clinical practice?
Sirukumab has not reached real-life clinical application, but the current evaluation of the long-term efficacy and particular safety of an IL-6 cytokine inhibitor in patients with RA is informative and relevant in a highly dynamic therapeutic field.
The data provided here may serve to further our understanding of different modes of action in RA.
Introduction
Rheumatoid arthritis (RA) is a chronic condition associated with high rates of pain, disability and comorbidity.1 Traditional treatment includes disease-modifying antirheumatic drugs (DMARDs), to which many patients do not respond or become intolerant.1 Therefore, compounds specifically involved in synovial inflammation such as tumour necrosis factor (TNF) and other proinflammatory pathway targets have been examined.
Interleukin (IL)-6 is a pleiotropic cytokine with proinflammatory functions, which include differentiation of B and helper T cells, and is involved in maintaining chronic inflammation in RA.2 Increased levels of IL-6, IL-1 and TNF have been observed in synovial fluid and serum in patients with RA.3 4
Sirukumab is a human monoclonal antibody directed against the IL-6 cytokine that inhibits IL-6-mediated signal transduction.5 6 In two phase III, randomised, double-blind, placebo-controlled, multicenter studies, sirukumab reduced signs and symptoms of RA, improved physical function and health status and inhibited radiographic progression in patients with moderately to severely active RA refractory to DMARDs (through 104 weeks; SIRROUND-D)6 7 or anti-TNF agents (through 52 weeks; SIRROUND-T).8 In SIRROUND-D, where radiographic progression was examined, sirukumab also inhibited radiographic progression.6 7
Since the sirukumab development programme was not further pursued based on a strategic decision by the sponsor, data from these studies and their extension will likely be the only evidence for long-term efficacy and safety of an IL-6 cytokine inhibitor. The primary objective of this study therefore was to assess and make available the long-term safety of sirukumab in patients with active RA refractory or intolerant to DMARDs or anti-TNF agents.
Methods
Study design
This was a multicenter, parallel-group, long-term extension (LTE) study of the SIRROUND-D and SIRROUND-T studies monitoring safety and efficacy of patients with moderately to severely active RA. The full methods and results of these studies have been published.6–8 Prior to early study termination, the original intention was to follow enrolled patients for 208 weeks. This study also included a substudy to assess usability of the SmartJect prefilled syringe autoinjector.
Patients
For inclusion in this study, patients completed the final study agent administrations from the primary studies (week 104 in SIRROUND-D and week 52 in SIRROUND-T) and provided informed consent. Patients were excluded if they had active diverticulitis or any condition that would compromise the well-being of the patient or preclude protocol-specified assessments.
For enrolment in SIRROUND-D and SIRROUND-T, patients were ≥18 years of age with moderately to severely active RA.6 8 For SIRROUND-D, patients were refractory to single-agent or combination DMARD therapy, including methotrexate or sulfasalazine, due to lack of benefit after ≥12 weeks as assessed by the treating physician. For SIRROUND-T, patients were refractory to previous anti-TNF agents due to an absence of benefit with ≥1 anti-TNF or documented intolerance to 1 anti-TNF. Patients were excluded if they had a history of intolerance to ≥2 (or inadequate response to ≥1) anti-TNF agents after 3 months or if they had received anti-TNF agents or other biologics for RA within the past 3 months.
Treatments
In SIRROUND-D and SIRROUND-T, patients were randomised 1:1:1 to sirukumab 50 mg every 4 weeks (q4w), sirukumab 100 mg every 2 weeks (q2w) or placebo delivered subcutaneously (SC). Patients in the placebo group were rerandomised at week 18 (early escape), week 24 (crossover in SIRROUND-T only), week 40 (late escape in SIRROUND-D) or at the crossover at week 52 (SIRROUND-D only).6 8 Eligible patients from primary studies remained on their blinded treatment, either sirukumab 100 mg SC q2w or sirukumab 50 mg SC q4w (with q4w placebo injections in between). Treatments were blinded until: (1) week 52 database lock occurred in SIRROUND-D, (2) week 24 database lock occurred in SIRROUND-T and (3) the last autoinjector usability substudy patient completed week 16 or was terminated from the study. Patients in the LTE study randomised in SIRROUND-D remained blinded until all three conditions occurred. Thereafter, the LTE became open-label; placebo injections were discontinued in the sirukumab 50 mg SC treatment group. Patients treated with sirukumab 100 mg q2w were offered the option to reduce the dose to 50 mg q4w during the open-label period, after discussion with their treating rheumatologist. Adverse events (AEs) occurring after patients had switched from the 100 mg q2w to the 50 mg q4w group were attributed to the 100 mg q2w group.
The maximum duration of participation in this study was to be 208 weeks of treatment, followed by approximately 16 weeks of safety and efficacy follow-up after the administration of the final injection of sirukumab. However, due to the sponsor’s decision to discontinue the sirukumab RA programme, the LTE was terminated early. Patients were scheduled for a final safety follow-up visit at study termination (31 December 2017).
Evaluations
Safety was based on reported AEs, clinical laboratory tests, vital sign measurements and physical examinations. An independent review committee assessed cases of major adverse cardiovascular events (MACE), and an independent gastroenterologist reviewed potential gastrointestinal perforation events. Documentation of events was subject based and not event based. Therefore, the main presentation of AEs is as per cent of subjects with event from enrolment in the LTE to last observation. However, to put the observed number of subjects with AEs in context and to allow comparison to other IL6-targeted compounds, we also calculated incidence rates per 100 patient-years (PY) for key treatment-emergent adverse event (TEAE) using the overall exposure time of each subject (ie, from first dose to last observation or first respective AE).
Efficacy was evaluated using the following assessments: tender and swollen joint count, patient’s and physician’s global assessment of disease activity, patient’s pain assessment, Health Assessment Questionnaire-Disability Index (HAQ-DI), serum C reactive protein (CRP) and duration of morning stiffness. These parameters were evaluated alone and/or in combination to assess other derived measures of efficacy including the American College of Rheumatology (ACR) response, Disease Activity Index Score 28 (CRP) (DAS28 (CRP)), ACR/EULAR remission, Clinical Disease Activity Index (CDAI) and Simplified Disease Activity Index (SDAI). No hypothesis tests were planned for this study.
Results
Patients
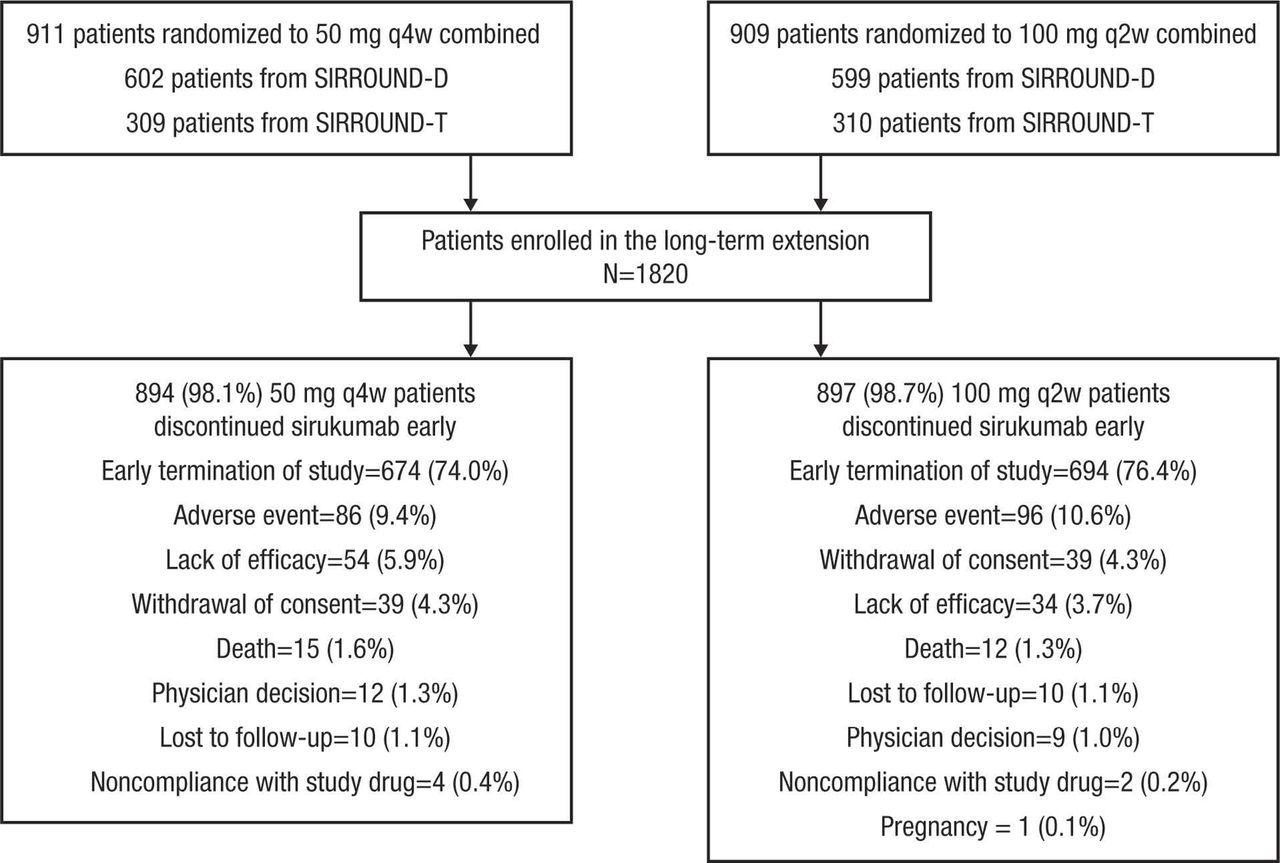
A total of 1820 patients were enrolled and included in the efficacy and safety analyses (figure 1). Of enrolled patients, 1368 (75.2%) discontinued sirukumab early due to the sponsor’s decision to terminate the study and further development of sirukumab for RA. Demographic and clinical characteristics were generally balanced across the sirukumab 50 mg q4w combined and 100 mg q2w combined groups (table 1).

Disposition of patients enrolling from SIRROUND-D and SIRROUND-T into SIRROUND-LTE. q2w, every 2 weeks; q4w, every 4 weeks.
Demographic and clinical characteristics at baseline in the primary studies*
Treatment
Median duration of exposure since the first LTE dose was 2.34 and 2.07 years in the sirukumab 50 mg q4w combined and sirukumab 100 mg q2w combined groups, respectively, and since first primary study dose was 3.78 and 3.83 years, respectively. Starting from the first LTE study dose, 1183/1820 (65.0%) patients had ≥104 weeks of sirukumab exposure; only 279/1820 (15.3%) patients achieved ≥156 weeks of sirukumab exposure. Overall, 29 patients completed the final on-treatment visit at week 208 before the sponsor’s decision to terminate the study. From the first dose in the primary studies, 80.3% of patients who enrolled in LTE achieved ≥156 weeks of sirukumab exposure.
Safety
Of 1820 patients enrolled, 1818 received ≥1 sirukumab dose. The two patients who did not receive sirukumab in the LTE were included in safety analyses because they received sirukumab in the primary studies, and there was no washout period between the primary studies and LTE.
An overall summary of AEs reported in patients after enrolling in the LTE is shown in table 2. AEs were reported in a similar proportion of patients in the sirukumab 50 mg q4w combined (78.7%) and 100 mg q2w combined (79.5%) groups, although non-similar rates were observed with injection-site reactions (3.7% in the sirukumab 50 mg q4w combined group and 7.5% in the sirukumab 100 mg q2w combined group) and MACE (2.2% in the sirukumab 50 mg q4w combined group and 1.0% in the sirukumab 100 mg q2w combined group). The most common serious AEs were pneumonia, cellulitis, osteoarthritis, sepsis and RA; the most common AEs resulting in study discontinuation were cellulitis, diverticulitis, pneumonia, sepsis and thrombocytopenia. Among these, there was a numerical trend indicative of a potential dose–response relationship particularly for pneumonia and sepsis, including septic shock (table 3).
Overall adverse events
Infections and infestations in the long-term extension
Overall, 27 patients in the combined sirukumab groups died during the primary studies. Overall, 32 (1.8%) patients died during the LTE study: 18 (2.0%) patients in the sirukumab 50 mg q4w combined group and 14 (1.5%) patients in the sirukumab 100 mg q2w combined group. Overall, 13 deaths were due to serious infection, 12 due to MACE and 1 due to malignancy. Other deaths were due to worsening chronic respiratory failure and diffuse interstitial lung disease, intestinal ischaemia as a result of diverticular perforation, mesenteric venous thrombosis and warfarin overdose (one patient each); two deaths were due to unknown causes.
Malignancies excluding non-melanoma skin cancer (NMSC) were reported by 24 (1.3%) patients: 9 (1.0%) patients in the sirukumab 50 mg q4w combined group (lymphoproliferative disorder (2 patients), acute promyelocytic leukaemia, colon cancer, myelofibrosis, papillary thyroid cancer, vaginal cancer, squamous cell carcinoma of pharynx, tonsil cancer) and 15 (1.6%) patients in the sirukumab 100 mg q2w group (breast cancer (5 patients), diffuse large B-cell lymphoma, malignant lung neoplasms (2 patients: 1 adenocarcinoma, 1 squamous cell carcinoma), malignant melanoma, bladder cancer, central nervous system lymphoma, non-Hodgkin’s lymphoma, rectal cancer, renal cancer, uterine cancer). One malignancy during the study period was fatal.
The incidence of hepatobiliary abnormalities was low, and no events of drug-induced liver injury occurred. Hepatobiliary abnormalities (detailed in table 2 footnote) were reported in three (0.2%) patients: one (0.1%) patient in the sirukumab 50 mg q4w combined group (increased alanine aminotransferase (ALT)≥3 times upper limit of normal (ULN)) and two (0.2%) patients in the sirukumab 100 mg q2w combined group (increased ALT (two events)≥3 times ULN and aspartate aminotransferase (AST)≥3 times ULN). One patient in the 100 mg q2w combined group had a grade 4 increase in ALT. No grade 4 toxicities were observed for AST or bilirubin.
Adjudicated MACE were reported in 29 (1.6%) patients (table 2), more had MACE in the sirukumab 50 mg q4w combined group (20 (2.2%)) compared with the 100 mg q2w combined group (9 (1.0%)). Myocardial infarctions were reported in six (0.3%) patients: three (0.3%) each in the 50 mg q4w and 100 mg q2w combined treatment groups. Strokes were reported in 13 (0.7%) patients: 8 (0.9%) in the 50 mg q4w combined group and 5 (0.5%) in the 100 mg q2w combined group. Other adjudicated MACE reported after enrolling in the LTE included unstable angina, cardiorespiratory arrest and sudden cardiac death in two patients each and transient ischaemic attack (in a patient who later developed a stroke), coronary artery insufficiency, death and subarachnoid haemorrhage in one patient each. Overall, 12 adjudicated cases of MACE were fatal. In addition, 14 (0.8%) patients had deep vein thrombosis or thrombosis.
Gastrointestinal perforations (all lower) occurred in 13 (0.7%) patients and were equally distributed among the treatment groups. Events in the 50 mg q4w combined group were: diverticular perforation, diverticulitis, intestinal ischaemia and appendicitis. Events in the 100 mg q2w combined group were: large intestine perforation and diverticulitis in one patient; large intestine perforation, intestinal obstruction, and diverticulitis in one patient; diverticulitis and colonic abscess in one patient; diverticulitis and colonic abscess in one patient; diverticular perforation in two patients and large intestine perforation and peritonitis. Two perforation events were fatal.
Overall, 102 (5.6%) patients had injection-site reactions, which were more frequent in the 100 mg q2w combined group compared with the 50 mg q4w combined group. Incidence of hypersensitivity reactions was low, with two (0.2%) patients in the 50 mg q4w combined group (hypersensitivity vasculitis and cutaneous vasculitis) and four (0.4%) patients in the 100 mg q2w combined group (urticaria (three patients) and hypersensitivity vasculitis). The only event considered serious was hypersensitivity vasculitis in the 100 mg q2w group, none were anaphylactic reactions.
The majority enrolled in the LTE had normal total cholesterol, low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C) and triglycerides values at primary study baseline. Among those with normal total cholesterol at baseline, 83.8% in the sirukumab 50 mg q4w combined group and 85.4% in the sirukumab 100 mg q2w combined group shifted from normal to a total cholesterol value >200 mg/dL. Two patients (one in each group) had grade 4 elevations in total cholesterol without discontinuation. Among patients with normal LDL-C at baseline, 69.9% in the sirukumab 50 mg q4w combined group and 69.9% in the sirukumab 100 mg q2w combined group shifted from normal to an LDL-C value >130 mg/dL by week 52 postbaseline. Of those with HDL-C values in the normal range at baseline, 16.1% in the sirukumab 50 mg q4w combined group and 15.6% in the sirukumab 100 mg q2w combined group had ≥1 HDL cholesterol values<40 mg/dL postbaseline. Among patients with normal fasting triglycerides values at baseline, 34.9% in the sirukumab 50 mg q4w combined group and 34.6% in the sirukumab 100 mg q2w combined group shifted from normal to a fasting triglyceride value >250 mg/dL. Six patients in the sirukumab 50 mg q4w combined group and seven in the sirukumab 100 mg q2w combined group had grade 4 elevations in triglycerides, without discontinuation. Patients were permitted to use statins for the treatment of hyperlipidemia as appropriate by National Cholesterol Education Program treatment guidelines or other national cholesterol treatment guidelines.
Haematologic abnormalities occurring since baseline of the primary studies through the end of the LTE were primarily grade 0 or 1 in severity across all treatment groups; only 10 patients had grade 4 events. Two patients had grade 4 lymphopenia (one in sirukumab 100 mg q2w group and one in the sirukumab 50 mg q4w combined group). Seven patients, all in the sirukumab 50 mg q4w combined group, experienced neutropenia. Of these patients, two discontinued from study agent. Sirukumab was interrupted in three patients and then resumed after neutrophil levels improved. One patient in the sirukumab 50 mg q4w group had leucopenia in addition to grade 4 neutropenia; the aetiology of these count drops was found to be leukaemia and sirukumab was discontinued. No grade 4 decreases were reported for platelets. Another patient with grade 4 neutropenia discontinued from study agent due to prolonged concomitant thrombocytopenia, although the thrombocytopenia did not reach grade 4.
Overall, 868 (47.7%) patients reported infections or infestations (table 3); similar incidences were observed in the sirukumab 50 mg q4w combined group (48.9%) and the sirukumab 100 mg q2w combined group (52.1%). The most common infections in the sirukumab combined group were viral upper respiratory tract infection (11.0%), upper respiratory tract infection (9.5%) and bronchitis (6.3%). Serious infections were reported in a similar proportion in the sirukumab 50 mg q4w combined group (9.6%) and the sirukumab 100 mg q2w combined group (11.1%). Overall, the most common serious infections were pneumonia (2.1%), cellulitis (1.5%) and sepsis (0.8%). Overall, 73 (4.0%) patients had treatment-emergent infections and infestations that led to discontinuation, the most common were cellulitis (0.4%), diverticulitis (0.4%), sepsis (0.4%) and osteomyelitis (0.3%).
Finally, online supplemental table 1 shows the subject-based incidence rates of key TEAE throughout the overall exposure to sirukumab. The overall incidence rates per 100 PY were 0.48 for deaths, 9.37 for serious AEs, 3.40 for serious infections and 0.59 for malignancies. Gastrointestinal perforations occurred at a rate of 0.19/100 PY and MACE at 0.56/100 PY (online supplemental table 1).
Supplemental material
Efficacy
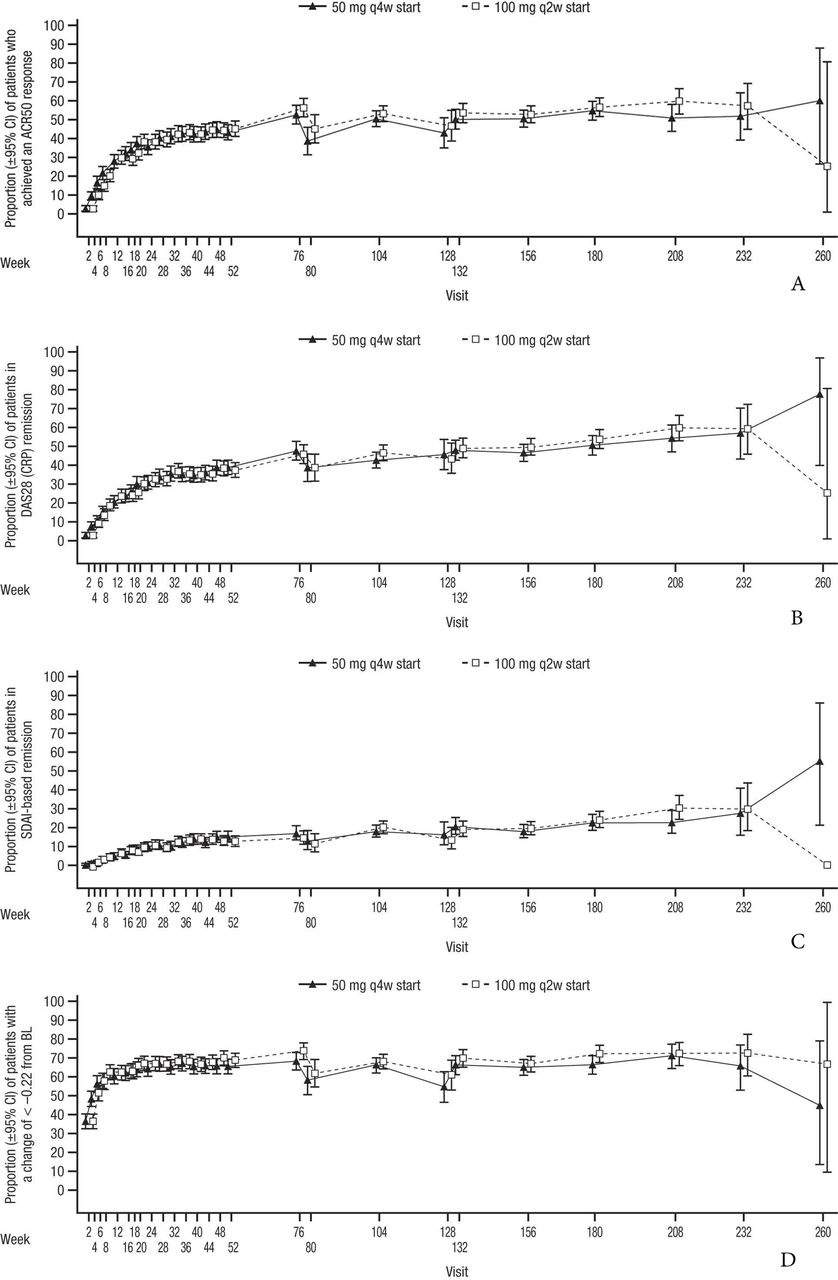
Of the patients enrolled in the LTE, the proportion achieving an ACR50 response (figure 2A), DAS28 (CRP) remission (figure 2B), SDAI-based ACR/EULAR remission (figure 2C) and HAQ-DI response (figure 2D) remained stable through the primary studies and during the SIRROUND-LTE study period, with a similar profile for both dosing regimens.

{kind=link}
{kind=link}
(A) Proportion of patients with ACR50 response from baseline of primary studies to the end of the SIRROUND-LTE study period. (B) Proportion of patients with DAS28 (CRP) remission from baseline of primary studies to the end of the SIRROUND-LTE study period. (C) Proportion of patients with SDAI-based ACR/EULAR remission from baseline of primary studies to the end of the SIRROUND-LTE study period. (D) Proportion of patients with HAQ-DI response from baseline of primary studies to the end of the SIRROUND-LTE study period. ACR, American College of Rheumatology; BL, baseline; DAS28 (CRP), Disease Activity Index Score 28 (C reactive protein); HAQ-DI, Health Assessment Questionnaire-Disability Index; LTE, long-term extension; q2w, every 2 weeks; q4w, every 4 weeks; SDAI, Simplified Disease Activity Index.
The proportion of patients who achieved ACR50 response was generally maintained and similar across both sirukumab dose groups. In the combined dose groups, 42.6%, 50.3% and 53.8% of evaluable patients demonstrated ACR50 response at weeks 52, 104 and 208 postbaseline, respectively. The proportion of patients who achieved Boolean-based ACR/EULAR remission (online supplemental figure 1A) and mean decreases in CDAI Score from baseline of primary studies (online supplemental figure 1B) were generally maintained during the SIRROUND-LTE study period and were similar in both sirukumab dose groups. In the combined dose groups, 7.8%, 11.2% and 14.7% of evaluable patients achieved ACR/EULAR remission at weeks 52, 104 and 208 postbaseline, respectively; evaluable patients achieved mean (SD) CDAI decreases of –22.99 (13.82), –25.93 (13.54) and –27.15 (13.22) at weeks 52, 104 and 208 postbaseline, respectively. Due to early study termination, efficacy data from only <2% of enrolled patients were available for the final on-treatment visit (week 260), leading to imprecise estimates as evidenced by the wide confidence intervals and should be interpreted with caution.
Discussion
This LTE study further evaluated the safety and efficacy of sirukumab, an IL-6 inhibitor, administered SC at doses of 50 mg q4w and 100 mg q2w in moderately to severely active RA refractory to DMARDs or anti-TNF agents. As the study was discontinued prematurely, only summary statistical analyses were conducted to evaluate long-term safety and laboratory parameters of interest. No new safety signals were detected with either sirukumab dose regimen. The improvements in symptoms of RA disease activity and physical function observed at the end of SIRROUND-D and SIRROUND-T were maintained for up to 2.3 years.
Overall, AEs were reported in a similar proportion of patients in the sirukumab 50 mg q4w and 100 mg q2w combined groups, and the safety profile was consistent with the primary studies.6–8 Injection-site reactions were more frequent in the sirukumab 100 mg q2w combined group, consistent with the primary studies,6–8 suggesting either a dose effect or an injection frequency effect. In addition, the long-term AE profile of sirukumab was also comparable to that of other agents blocking the IL-6 pathway, such as IL-6 receptor blockers sarilumab and tocilizumab. In a phase IIIb, 2-year LTE study for patients treated with tocilizumab, injection-site reactions occurred in 2.8% of patients.9 In the sarilumab LTE study, of 2887 patients on combination therapy with conventional synthetic DMARDs (up to 7.3 years of exposure), 7.5% reported injection-site reactions (13.3/100 PY), and of 471 patients on monotherapy (up to 3.5 years exposure), 8.1% reported injection-site reactions (25.6/100 PY).10
A total of 32 deaths were reported in the study (27 in the primary study periods and 5 in the LTE). The causes of death were primarily due to serious infections and MACE, which are the leading causes of death in the RA population.11–13 Considering the death/100 PY, the rates correspond to 0.5 (50 mg q4w) and 0.4 (100 mg q2w). By comparison, in a cumulative analysis of long-term tocilizumab data in RA (up to 4.6 years of exposure), of 4009 patients in the all-exposed population, 55 deaths (0.45/100 PY) were reported occurring in ≤84 days after the last dose14; in the sarilumab LTE, 31 deaths occurred out of 2887 patients on combination therapy (0.4/100 PY) and 5 deaths occurred out of 471 patients on monotherapy (0.6/100 PY).10
Nine non-NMSC malignancies were reported in the sirukumab 50 mg q4w combined group and fifteen were reported in the sirukumab 100 mg q2w combined group. Overall, the number and type of malignancies in this study were consistent with malignancies in patients with RA.15–17 In the tocilizumab LTE study, malignancies occurred at a rate of 1.2/100 PY, and for sarilumab, malignancies occurred at 0.7/100 PY (combination) and 0.6/100 PY (monotherapy), respectively.10 14
Infections, MACE, alterations in serum lipids and gastrointestinal perforations are all known AEs associated with IL-6 inhibitors; these AEs are consistent with the role of IL-6 in immunity, tissue homeostasis and cardiovascular disease.2 The most frequent types of AE in the LTE were infections or infestations, which occurred in almost half of patients. Approximately 10% of all patients reported serious infections and 45 (2.5%) patients had herpes zoster over approximately 2 years of sirukumab exposure in the LTE. By comparison, rates of serious infections reported were 4.5/100 PY for tocilizumab and 3.7/100 PY and 1.0/100 PY for sarilumab combination and monotherapy, respectively.10 14 For serious pneumonia, the rates on sirukumab were 0.7 (50 mg q4w) and 1.0 (100mg q2w), compared with 0.7 and 0.6 for tocilizumab and sarilumab, respectively.
MACE occurred in 29 (1.9%) patients on sirukumab, two times as frequently in the 50 mg q4w group compared with the 100 mg q2w group; deep vein thrombosis or thrombosis were observed in 0.8% of all patients. Cardiovascular disease is prevalent in patients with RA and other inflammatory conditions.15 18 There was no apparent imbalance in the combined 50 versus 100 mg groups for MACE risk factors; thus, it remains unclear why the MACE rate in the 50 mg q4w group was two times that of the 100 mg q2w group (2.2% vs 1.0%). For tocilizumab, the rate of myocardial infarction was 0.29/100 PY,14 and for sarilumab, incidences of MACE were 1.4% (combination; 0.5/100 PY) and 0.4% (monotherapy; 0.2/100 PY).10 The sarilumab study also reported deep vein thrombosis rates of 0.2/100 PY (combination) and 0.1/100 PY (monotherapy).
Laboratory abnormalities including neutropenia, thrombocytopenia and increased levels of transaminases and lipid panels were associated with sirukumab, all of which have been reported as class effects of anti-IL-6 agents in previous clinical studies.9 17 19 The majority of ALT and AST elevations were mild and asymptomatic. Increases in total cholesterol, LDL-C, HDL-C and fasting triglycerides were observed with sirukumab treatment. This is consistent with an observed increase in lipid parameters associated with other anti-IL-6 agents.19 As with the entire class of anti-IL-6 agents, the association of dyslipidaemia with risk of cardiovascular disease needs further research.
Gastrointestinal perforations were rare and occurred in 13 patients. Gastrointestinal perforations are a known consequence of anti-IL-6 therapy19 and the events reported in this study (0.2/100 PY) are consistent with prior tocilizumab and sarilumab studies (0.2 and 0.1 per 100 PY, respectively).10 14
The strengths of this study include the large patient population and extended exposure time to sirukumab, which was up to 5 years for some patients. Furthermore, enrolled patients had received both non-biologic and biologic DMARDs as prior therapies, representing a difficult to treat population. This study is limited by the lack of a long-term placebo comparator; thus, it cannot be known with certainty which events are due to sirukumab. The premature end to the study also limited the total number of patients who contributed to some of the long-term efficacy endpoints.
In conclusion, this study found that the long-term safety of sirukumab was consistent with that of the primary studies and with other IL-6 pathway inhibitors in this established RA population. Sirukumab also maintained the reduction in clinical signs and symptoms of RA and improvements in physical function achieved at the completion of the primary studies. Although the compound has not reached real-life clinical application, the value of this evaluation based on a large clinical programme of an IL-6 cytokine inhibitor in RA is informative and relevant in a highly dynamic therapeutic field and to further our understanding of different modes of action in RA.
Acknowledgments
Writing and editorial support were provided by Allison Michaelis, PhD, MedErgy.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors DA, COB, GAK, TT, CT and YT were involved in design of the analyses, data interpretation and write up/editing of the manuscript. In addition, AB, PA, BH, RR and KB were involved or partly involved in data acquisition.
Funding This study was sponsored by Janssen Research & Development, LLC, in collaboration with GlaxoSmithKline.
Competing interests DA has received consulting and/or speaking fees from AbbVie, Amgen, Celgene, Gilead, Galappagos, Lilly, Medac, Merck, Novartis, Pfizer, Roche, Sandoz and Sanofi/Genzyme. COB has received consulting fees from AbbVie, Bristol-Myers Squibb, Eli Lilly, Genentech/Roche, Janssen, Pfizer and Regeneron/Sanofi and grant support from Bristol-Myers Squibb and Janssen. CT has received consulting and/or advisory board fees from AbbVie, Amgen, AstraZeneca, BMS, Celgene, CaREBiodam, Centocor, Genzyme, GSK, Hospira, Janssen, Lilly, Medexus/Medac, Merck, Novartis, Pfizer, Roche, Sandoz, Sanofi, Takeda, TcLand and UCB and speaker fees from Medexus/Medac. GAK has received advisory and/or speaking fees from Bristol-Myers Squibb, Sanofi-Genzyme Regeneron and Janssen and grant support from Pfizer. TT has received grants from Astellas, Chugai, Daiichi Sankyo, Takeda, AbbVie GK, Asahi Kasei, Mitsubishi Tanabe, Eisai, Nippon Kayaku and JCR, speaking fees and/or honoraria from AbbVie GK, Chugai, Bristol-Myers KK, Eli Lilly Japan KK, SRL, Astellas, AYUMI, Eisai, Ono, Kissei, Gilead Sciences, Mitsubishi Tanabe, Novartis Pharma KK, Pfizer Japan, Taiho, Daiichi Sankyo, Taisho, Nippon Kayaku, Boehringer Ingelheim, A2 Healthcare and The Uehara Memorial Foundation and consulting fees from Astellas, AbbVie GK, Ono, Gilead Sciences, Taiho, Daiichi Sankyo, Taisho, Nippon Kayaku, Bristol-Myers KK, Chugai, Novartis Pharma KK, A2 Healthcare Corporation and Boehringer Ingelheim. AB was an employee at Janssen when the study was performed and is currently an employee at Merck. PA is an employee of Janssen Pharmaceuticals. BH was an employee at Janssen when the study was performed. RR was an employee at GlaxoSmithKline when the study was performed. KB was an employee at GlaxoSmithKline when the study was performed and is currently an employee at Incyte. YT has received speaking fees and/or honoraria from Daiichi-Sankyo, Astellas, Chugai, Eli Lilly, Pfizer, AbbVie, YL Biologics, Bristol-Myers, Takeda, Mitsubishi-Tanabe, Novartis, Eisai, Janssen and Teijin and research grants from Asahi Kasei, Mitsubishi-Tanabe, Chugai, Takeda, Sanofi, Bristol-Myers, UCB, Daiichi-Sankyo, Eisai and Ono.
Patient consent for publication Not required.
Ethics approval Not applicable: secondary analysis of completed programmes. Consent provided to original studies.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement No data are available.